当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Electronic Excitations in Crystalline Solids through the Maximum Overlap Method
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2021-09-30 , DOI: 10.1021/acs.jctc.1c00427 Loredana Edith Daga 1 , Lorenzo Maschio 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2021-09-30 , DOI: 10.1021/acs.jctc.1c00427 Loredana Edith Daga 1 , Lorenzo Maschio 1
Affiliation
|
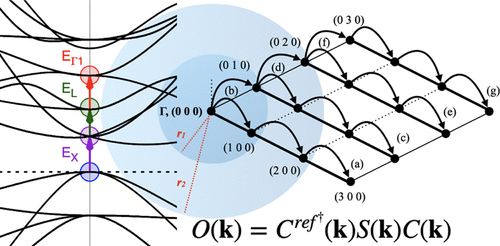
The maximum overlap method (MOM) has emerged from molecular quantum chemistry as a convenient practical procedure for studying excited states. Unlike the Aufbau principle, during self-consistent field (SCF) iterations, the MOM forces orbital occupation to be maximally similar to that of a reference state. Although still within a single-particle framework, this approach allows for the evaluation of excitation energies (Δ-SCF) and geometry optimization of electronic configurations other than the ground state. In this work, we present an extension of the MOM to periodic crystalline solids, within the framework of an atom-centered Gaussian basis set. In order to obtain a realistic concentration of excited electrons, we allow excitation in only one—or a few—points of the Brillouin zone, leading to a fractional occupation of crystalline Kohn–Sham states. Since periodic SCF solution techniques involve an iteration between direct and reciprocal spaces, only totally symmetric excitations are allowed in our treatment, in order to preserve the translational symmetry: vertical Γ-point excitations or collective excitations in a sphere around Γ. Other types of excitations are accessible through folding of the Brillouin zone subsequent to the creation of a supercell. The features and performance of the method are presented through its application to prototypical solids such as bulk silicon, diamond, and lithium fluoride and comparing the results with the available experimental data. The demonstrative application to nickel oxide and solid CuI(piperazine)—a luminescent copper halide compound—highlights the promising potential of the MOM in solid-state quantum chemistry.
中文翻译:

通过最大重叠法在结晶固体中进行电子激发
最大重叠法 (MOM) 已从分子量子化学中出现,作为研究激发态的一种方便实用的方法。与 Aufbau 原理不同,在自洽场 (SCF) 迭代期间,MOM 强制轨道占用与参考状态的轨道占用最大程度地相似。尽管仍处于单粒子框架内,但这种方法允许评估激发能 (Δ-SCF) 和基态以外的电子配置的几何优化。在这项工作中,我们在以原子为中心的高斯基组的框架内,将 MOM 扩展到周期性结晶固体。为了获得真实的激发电子浓度,我们只允许在布里渊区的一个或几个点进行激发,从而导致部分占据晶体 Kohn-Sham 态。由于周期性 SCF 求解技术涉及直接空间和倒易空间之间的迭代,因此在我们的处理中只允许完全对称的激发,以保持平移对称性:垂直 Γ 点激发或围绕 Γ 的球体中的集体激发。其他类型的激发可以通过在超级单体产生之后折叠布里渊区来获得。该方法的特点和性能通过将其应用于原型固体(如块状硅、金刚石和氟化锂)并将结果与可用的实验数据进行比较来介绍。对氧化镍和固体 CuI(哌嗪)(一种发光的卤化铜化合物)的示范应用凸显了 MOM 在固态量子化学中的潜力。
更新日期:2021-10-12
中文翻译:
通过最大重叠法在结晶固体中进行电子激发
最大重叠法 (MOM) 已从分子量子化学中出现,作为研究激发态的一种方便实用的方法。与 Aufbau 原理不同,在自洽场 (SCF) 迭代期间,MOM 强制轨道占用与参考状态的轨道占用最大程度地相似。尽管仍处于单粒子框架内,但这种方法允许评估激发能 (Δ-SCF) 和基态以外的电子配置的几何优化。在这项工作中,我们在以原子为中心的高斯基组的框架内,将 MOM 扩展到周期性结晶固体。为了获得真实的激发电子浓度,我们只允许在布里渊区的一个或几个点进行激发,从而导致部分占据晶体 Kohn-Sham 态。由于周期性 SCF 求解技术涉及直接空间和倒易空间之间的迭代,因此在我们的处理中只允许完全对称的激发,以保持平移对称性:垂直 Γ 点激发或围绕 Γ 的球体中的集体激发。其他类型的激发可以通过在超级单体产生之后折叠布里渊区来获得。该方法的特点和性能通过将其应用于原型固体(如块状硅、金刚石和氟化锂)并将结果与可用的实验数据进行比较来介绍。对氧化镍和固体 CuI(哌嗪)(一种发光的卤化铜化合物)的示范应用凸显了 MOM 在固态量子化学中的潜力。










































 京公网安备 11010802027423号
京公网安备 11010802027423号