当前位置:
X-MOL 学术
›
Chem. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
A generic approach to decipher the mechanistic pathway of heterogeneous protein aggregation kinetics
Chemical Science ( IF 7.6 ) Pub Date : 2021-09-03 , DOI: 10.1039/d1sc03190b Baishakhi Tikader 1 , Samir K Maji 2 , Sandip Kar 1
Chemical Science ( IF 7.6 ) Pub Date : 2021-09-03 , DOI: 10.1039/d1sc03190b Baishakhi Tikader 1 , Samir K Maji 2 , Sandip Kar 1
Affiliation
|
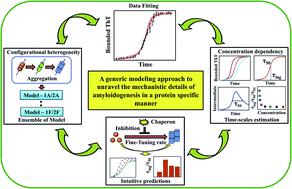
Amyloid formation is a generic property of many protein/polypeptide chains. A broad spectrum of proteins, despite having diversity in the inherent precursor sequence and heterogeneity present in the mechanism of aggregation produces a common cross β-spine structure that is often associated with several human diseases. However, a general modeling framework to interpret amyloid formation remains elusive. Herein, we propose a data-driven mathematical modeling approach that elucidates the most probable interaction network for the aggregation of a group of proteins (α-synuclein, Aβ42, Myb, and TTR proteins) by considering an ensemble set of network models, which include most of the mechanistic complexities and heterogeneities related to amyloidogenesis. The best-fitting model efficiently quantifies various timescales involved in the process of amyloidogenesis and explains the mechanistic basis of the monomer concentration dependency of amyloid-forming kinetics. Moreover, the present model reconciles several mutant studies and inhibitor experiments for the respective proteins, making experimentally feasible non-intuitive predictions, and provides further insights about how to fine-tune the various microscopic events related to amyloid formation kinetics. This might have an application to formulate better therapeutic measures in the future to counter unwanted amyloidogenesis. Importantly, the theoretical method used here is quite general and can be extended for any amyloid-forming protein.
中文翻译:

破译异质蛋白质聚集动力学机制途径的通用方法
淀粉样蛋白的形成是许多蛋白质/多肽链的通用特性。尽管固有前体序列具有多样性并且聚集机制存在异质性,但多种蛋白质产生了常见的交叉β-脊柱结构,该结构通常与多种人类疾病相关。然而,解释淀粉样蛋白形成的通用模型框架仍然难以捉摸。在此,我们提出了一种数据驱动的数学建模方法,通过考虑一组网络模型来阐明一组蛋白质(α-突触核蛋白、Aβ42、Myb 和 TTR 蛋白质)聚集的最可能的相互作用网络,其中包括大多数机制的复杂性和异质性与淀粉样蛋白生成有关。最佳拟合模型有效地量化了淀粉样蛋白生成过程中涉及的各种时间尺度,并解释了淀粉样蛋白形成动力学的单体浓度依赖性的机制基础。此外,本模型协调了各自蛋白质的多项突变研究和抑制剂实验,做出了实验上可行的非直观预测,并提供了有关如何微调与淀粉样蛋白形成动力学相关的各种微观事件的进一步见解。这可能有助于在未来制定更好的治疗措施来对抗不必要的淀粉样蛋白生成。重要的是,这里使用的理论方法非常通用,可以扩展到任何淀粉样蛋白形成蛋白。
更新日期:2021-09-28
中文翻译:
破译异质蛋白质聚集动力学机制途径的通用方法
淀粉样蛋白的形成是许多蛋白质/多肽链的通用特性。尽管固有前体序列具有多样性并且聚集机制存在异质性,但多种蛋白质产生了常见的交叉β-脊柱结构,该结构通常与多种人类疾病相关。然而,解释淀粉样蛋白形成的通用模型框架仍然难以捉摸。在此,我们提出了一种数据驱动的数学建模方法,通过考虑一组网络模型来阐明一组蛋白质(α-突触核蛋白、Aβ42、Myb 和 TTR 蛋白质)聚集的最可能的相互作用网络,其中包括大多数机制的复杂性和异质性与淀粉样蛋白生成有关。最佳拟合模型有效地量化了淀粉样蛋白生成过程中涉及的各种时间尺度,并解释了淀粉样蛋白形成动力学的单体浓度依赖性的机制基础。此外,本模型协调了各自蛋白质的多项突变研究和抑制剂实验,做出了实验上可行的非直观预测,并提供了有关如何微调与淀粉样蛋白形成动力学相关的各种微观事件的进一步见解。这可能有助于在未来制定更好的治疗措施来对抗不必要的淀粉样蛋白生成。重要的是,这里使用的理论方法非常通用,可以扩展到任何淀粉样蛋白形成蛋白。










































 京公网安备 11010802027423号
京公网安备 11010802027423号