当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Machine Learning-Assisted Selection of Active Spaces for Strongly Correlated Transition Metal Systems
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2021-09-27 , DOI: 10.1021/acs.jctc.1c00235 Pavlo Golub 1 , Andrej Antalik 1 , Libor Veis 1 , Jiri Brabec 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2021-09-27 , DOI: 10.1021/acs.jctc.1c00235 Pavlo Golub 1 , Andrej Antalik 1 , Libor Veis 1 , Jiri Brabec 1
Affiliation
|
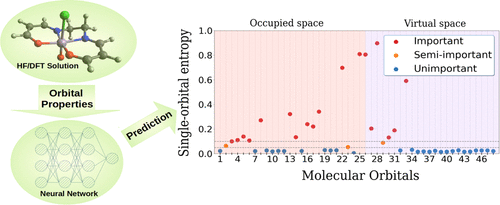
Active space quantum chemical methods could provide very accurate description of strongly correlated electronic systems, which is of tremendous value for natural sciences. The proper choice of the active space is crucial but a nontrivial task. In this article, we present a neural network-based approach for automatic selection of active spaces, focused on transition metal systems. The training set has been formed from artificial systems composed of one transition metal and various ligands, on which we have performed the density matrix renormalization group and calculated the single-site entropy. On the selected set of systems, ranging from small benchmark molecules up to larger challenging systems involving two metallic centers, we demonstrate that our machine learning models could predict the active space orbitals with reasonable accuracy. We also tested the transferability on out-of-the-model systems, including bimetallic complexes and complexes with ligands, which were not involved in the training set. Also, we tested the correctness of the automatically selected active spaces on a Fe(II)–porphyrin model, where we studied the lowest states at the DMRG level and compared the energy difference between spin states or the energy difference between conformations of ferrocene with recent studies.
中文翻译:

强相关过渡金属系统的活动空间的机器学习辅助选择
主动空间量子化学方法可以非常准确地描述强相关电子系统,这对自然科学具有巨大的价值。正确选择活动空间是至关重要的,但也是一项重要的任务。在本文中,我们提出了一种基于神经网络的自动选择活动空间的方法,重点是过渡金属系统。训练集由一种过渡金属和各种配体组成的人工系统组成,我们对其进行了密度矩阵重整化组并计算了单点熵。在选定的系统集上,从小型基准分子到涉及两个金属中心的更大具有挑战性的系统,我们证明了我们的机器学习模型可以以合理的精度预测活动空间轨道。我们还测试了模型外系统的可转移性,包括双金属配合物和配体配合物,这些都没有参与训练集。此外,我们在 Fe(II)-卟啉模型上测试了自动选择的活性空间的正确性,我们研究了 DMRG 水平的最低状态,并将自旋态之间的能量差异或二茂铁构象之间的能量差异与最近的学习。
更新日期:2021-10-12
中文翻译:
强相关过渡金属系统的活动空间的机器学习辅助选择
主动空间量子化学方法可以非常准确地描述强相关电子系统,这对自然科学具有巨大的价值。正确选择活动空间是至关重要的,但也是一项重要的任务。在本文中,我们提出了一种基于神经网络的自动选择活动空间的方法,重点是过渡金属系统。训练集由一种过渡金属和各种配体组成的人工系统组成,我们对其进行了密度矩阵重整化组并计算了单点熵。在选定的系统集上,从小型基准分子到涉及两个金属中心的更大具有挑战性的系统,我们证明了我们的机器学习模型可以以合理的精度预测活动空间轨道。我们还测试了模型外系统的可转移性,包括双金属配合物和配体配合物,这些都没有参与训练集。此外,我们在 Fe(II)-卟啉模型上测试了自动选择的活性空间的正确性,我们研究了 DMRG 水平的最低状态,并将自旋态之间的能量差异或二茂铁构象之间的能量差异与最近的学习。










































 京公网安备 11010802027423号
京公网安备 11010802027423号