当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Kinematic Reconstruction of Cyclic Peptides and Protein Backbones from Partial Data
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2021-09-27 , DOI: 10.1021/acs.jcim.1c00453 Mosavverul Hassan 1 , Evangelos A Coutsias 1, 2
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2021-09-27 , DOI: 10.1021/acs.jcim.1c00453 Mosavverul Hassan 1 , Evangelos A Coutsias 1, 2
Affiliation
|
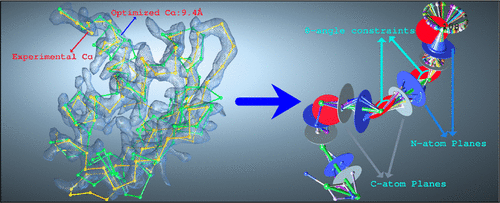
We present an algorithm, QBKR (Quaternary Backbone Kinematic Reconstruction), a fast analytical method for an all-atom backbone reconstruction of proteins and linear or cyclic peptide chains from Cα coordinate traces. Unlike previous analytical methods for deriving all-atom representations from coarse-grained models that rely on canonical geometry with planar peptides in the trans conformation, our de novo kinematic model incorporates noncanonical, cis–trans, geometry naturally. Perturbations to this geometry can be effected with ease in our formulation, for example, to account for a continuous change from cis to trans geometry. A simple optimization of a spring-based objective function is employed for Cα–Cα distance variations that extend beyond the cis–trans limit. The kinematic construction produces a linked chain of peptide units, Cα–C–N–Cα, hinged at the Cα atoms spanning all possible planar and nonplanar peptide conformations. We have combined our method with a ring closure algorithm for the case of ring peptides and missing loops in a protein structure. Here, the reconstruction proceeding from both the N and C termini of the protein backbone (or in both directions from a starting position for rings) requires freedom in the position of one Cα atom (a capstone) to achieve a successful loop or ring closure. A salient feature of our reconstruction method is the ability to enrich conformational ensembles to produce alternative feasible conformations in which H-bond forming C–O or N–H pairs in the backbone can reverse orientations, thus addressing a well-known shortcoming in Cα-based RMSD structure comparison, wherein very close structures may lead to significantly different overall H-bond behavior. We apply the fixed Cα-based design to the reverse reconstruction from noisy Cryo-EM data, a posteriori to the optimization. Our method can be applied to speed up the process of an all-atom description from voluminous experimental data or subpar electron density maps.
中文翻译:

从部分数据重建环肽和蛋白质骨架的运动学
我们提出了一种算法 QBKR(第四纪骨干运动学重建),这是一种快速分析方法,用于从C α坐标迹线重建蛋白质和线性或环状肽链的全原子骨干。与以前的分析方法不同,这些方法从依赖于反式构象平面肽的规范几何的粗粒度模型导出全原子表示,我们的从头运动学模型自然地结合了非规范、顺式-反式几何。在我们的公式中可以很容易地对这种几何结构进行扰动,例如,以解释从顺式到反式的连续变化几何学。基于弹簧的目标函数的简单优化用于C α - C α距离变化超出顺式-反式限制。运动学构造产生肽单元的连接链,C α – C – N – C α,铰接在跨越所有可能的平面和非平面肽构象的C α原子上。我们将我们的方法与闭环算法相结合,以解决蛋白质结构中环肽和缺失环的情况。在这里,从N和C开始的重建蛋白质主链的末端(或环起始位置的两个方向)需要一个C α原子(顶点)的位置自由,以实现成功的环或环闭合。我们的重建方法的一个显着特征是能够丰富构象集合以产生替代的可行构象,其中在骨架中形成 C-O 或 N-H 对的氢键可以反转方向,从而解决C α中众所周知的缺点- 基于 RMSD 结构比较,其中非常接近的结构可能导致显着不同的整体氢键行为。我们应用固定的C α基于设计到从嘈杂的 Cryo-EM 数据进行逆向重建,这是优化的后验。我们的方法可用于加速从大量实验数据或低于标准的电子密度图进行全原子描述的过程。
更新日期:2021-10-25
中文翻译:
从部分数据重建环肽和蛋白质骨架的运动学
我们提出了一种算法 QBKR(第四纪骨干运动学重建),这是一种快速分析方法,用于从C α坐标迹线重建蛋白质和线性或环状肽链的全原子骨干。与以前的分析方法不同,这些方法从依赖于反式构象平面肽的规范几何的粗粒度模型导出全原子表示,我们的从头运动学模型自然地结合了非规范、顺式-反式几何。在我们的公式中可以很容易地对这种几何结构进行扰动,例如,以解释从顺式到反式的连续变化几何学。基于弹簧的目标函数的简单优化用于C α - C α距离变化超出顺式-反式限制。运动学构造产生肽单元的连接链,C α – C – N – C α,铰接在跨越所有可能的平面和非平面肽构象的C α原子上。我们将我们的方法与闭环算法相结合,以解决蛋白质结构中环肽和缺失环的情况。在这里,从N和C开始的重建蛋白质主链的末端(或环起始位置的两个方向)需要一个C α原子(顶点)的位置自由,以实现成功的环或环闭合。我们的重建方法的一个显着特征是能够丰富构象集合以产生替代的可行构象,其中在骨架中形成 C-O 或 N-H 对的氢键可以反转方向,从而解决C α中众所周知的缺点- 基于 RMSD 结构比较,其中非常接近的结构可能导致显着不同的整体氢键行为。我们应用固定的C α基于设计到从嘈杂的 Cryo-EM 数据进行逆向重建,这是优化的后验。我们的方法可用于加速从大量实验数据或低于标准的电子密度图进行全原子描述的过程。








































 京公网安备 11010802027423号
京公网安备 11010802027423号