当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Theoretical Prediction and Interpretation of 237Np Mössbauer Isomer Shifts
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2021-09-22 , DOI: 10.1021/acs.jctc.1c00687 Laura C Motta 1 , Jochen Autschbach 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2021-09-22 , DOI: 10.1021/acs.jctc.1c00687 Laura C Motta 1 , Jochen Autschbach 1
Affiliation
|
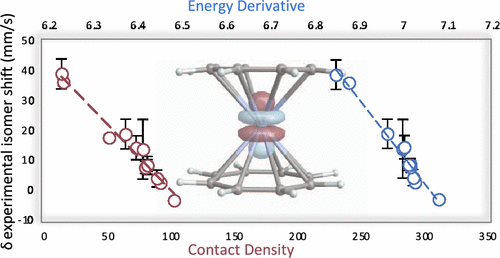
Different theoretical approaches for the calculation of 237Np Mössbauer isomer shifts are investigated. The traditional contact density route is compared to a previously proposed alternative approach that uses energy derivatives with respect to the nuclear radius. Both approaches yield similar results as long as suitable basis sets augmented with large exponents and relativistic methods are used. Density functional theory (DFT) calculations do not show a strong dependency of the 237Np isomer shift on the chosen functional. Wavefunction calculations show that dynamic electron correlation can be important when covalent bonding influences the isomer shift. Effects from spin–orbit coupling are small. The isomer shifts of ionic solids and Np(III) organometallic complexes are largely governed by the oxidation state of Np. Isomer shifts of organometallic Np(IV) complexes are strongly affected by donation bonding. Detailed analysis of the wavefunction results with different active spaces demonstrates that correlation among the outer core Np and occupied ligand frontier orbitals contributes significantly to isomer shifts of Np(IV) compounds.
中文翻译:

237Np Mössbauer 异构体位移的理论预测和解释
研究了计算237 Np Mössbauer 异构体位移的不同理论方法。将传统的接触密度路线与先前提出的使用相对于核半径的能量导数的替代方法进行比较。只要使用用大指数和相对论方法增强的合适基组,这两种方法都会产生相似的结果。密度泛函理论 (DFT) 计算未显示237的强依赖性所选功能的 Np 异构体转移。波函数计算表明,当共价键合影响异构体转移时,动态电子相关性可能很重要。自旋轨道耦合的影响很小。离子固体和 Np(III) 有机金属配合物的异构体转移主要由 Np 的氧化态决定。有机金属 Np(IV) 配合物的异构体转移受捐赠键合的强烈影响。对具有不同活性空间的波函数结果的详细分析表明,外核 Np 和已占配体前沿轨道之间的相关性对 Np(IV) 化合物的异构体移动有显着影响。
更新日期:2021-10-12
中文翻译:
237Np Mössbauer 异构体位移的理论预测和解释
研究了计算237 Np Mössbauer 异构体位移的不同理论方法。将传统的接触密度路线与先前提出的使用相对于核半径的能量导数的替代方法进行比较。只要使用用大指数和相对论方法增强的合适基组,这两种方法都会产生相似的结果。密度泛函理论 (DFT) 计算未显示237的强依赖性所选功能的 Np 异构体转移。波函数计算表明,当共价键合影响异构体转移时,动态电子相关性可能很重要。自旋轨道耦合的影响很小。离子固体和 Np(III) 有机金属配合物的异构体转移主要由 Np 的氧化态决定。有机金属 Np(IV) 配合物的异构体转移受捐赠键合的强烈影响。对具有不同活性空间的波函数结果的详细分析表明,外核 Np 和已占配体前沿轨道之间的相关性对 Np(IV) 化合物的异构体移动有显着影响。










































 京公网安备 11010802027423号
京公网安备 11010802027423号