当前位置:
X-MOL 学术
›
Chem. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Structure-based de novo drug design using 3D deep generative models
Chemical Science ( IF 7.6 ) Pub Date : 2021-09-09 , DOI: 10.1039/d1sc04444c Yibo Li 1 , Jianfeng Pei 2 , Luhua Lai 1, 2, 3
Chemical Science ( IF 7.6 ) Pub Date : 2021-09-09 , DOI: 10.1039/d1sc04444c Yibo Li 1 , Jianfeng Pei 2 , Luhua Lai 1, 2, 3
Affiliation
|
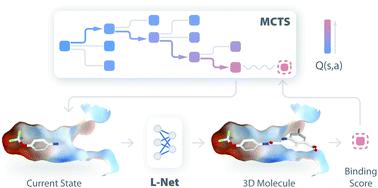
Deep generative models are attracting much attention in the field of de novo molecule design. Compared to traditional methods, deep generative models can be trained in a fully data-driven way with little requirement for expert knowledge. Although many models have been developed to generate 1D and 2D molecular structures, 3D molecule generation is less explored, and the direct design of drug-like molecules inside target binding sites remains challenging. In this work, we introduce DeepLigBuilder, a novel deep learning-based method for de novo drug design that generates 3D molecular structures in the binding sites of target proteins. We first developed Ligand Neural Network (L-Net), a novel graph generative model for the end-to-end design of chemically and conformationally valid 3D molecules with high drug-likeness. Then, we combined L-Net with Monte Carlo tree search to perform structure-based de novo drug design tasks. In the case study of inhibitor design for the main protease of SARS-CoV-2, DeepLigBuilder suggested a list of drug-like compounds with novel chemical structures, high predicted affinity, and similar binding features to those of known inhibitors. The current version of L-Net was trained on drug-like compounds from ChEMBL, which could be easily extended to other molecular datasets with desired properties based on users' demands and applied in functional molecule generation. Merging deep generative models with atomic-level interaction evaluation, DeepLigBuilder provides a state-of-the-art model for structure-based de novo drug design and lead optimization.
中文翻译:

使用 3D 深度生成模型的基于结构的从头药物设计
深度生成模型在从头分子设计领域备受关注。与传统方法相比,深度生成模型可以以完全数据驱动的方式进行训练,几乎不需要专家知识。尽管已经开发了许多模型来生成 1D 和 2D 分子结构,但对 3D 分子生成的探索较少,并且在目标结合位点内直接设计类药物分子仍然具有挑战性。在这项工作中,我们介绍了 DeepLigBuilder,这是一种新的基于深度学习的de novo方法在目标蛋白的结合位点生成 3D 分子结构的药物设计。我们首先开发了配体神经网络 (L-Net),这是一种新颖的图形生成模型,用于端到端设计具有高药物相似性的化学和构象有效 3D 分子。然后,我们结合 L-Net 和 Monte Carlo 树搜索来执行基于结构的de novo药物设计任务。在 SARS-CoV-2 主要蛋白酶抑制剂设计的案例研究中,DeepLigBuilder 提出了一系列具有新化学结构、高预测亲和力和与已知抑制剂相似的结合特征的类药物化合物。当前版本的 L-Net 是针对来自 ChEMBL 的类药物化合物进行训练的,可以根据用户的需求轻松地将其扩展到具有所需特性的其他分子数据集,并应用于功能分子的生成。DeepLigBuilder 将深度生成模型与原子级相互作用评估相结合,为基于结构的从头药物设计和先导优化提供了最先进的模型。
更新日期:2021-09-22
中文翻译:
使用 3D 深度生成模型的基于结构的从头药物设计
深度生成模型在从头分子设计领域备受关注。与传统方法相比,深度生成模型可以以完全数据驱动的方式进行训练,几乎不需要专家知识。尽管已经开发了许多模型来生成 1D 和 2D 分子结构,但对 3D 分子生成的探索较少,并且在目标结合位点内直接设计类药物分子仍然具有挑战性。在这项工作中,我们介绍了 DeepLigBuilder,这是一种新的基于深度学习的de novo方法在目标蛋白的结合位点生成 3D 分子结构的药物设计。我们首先开发了配体神经网络 (L-Net),这是一种新颖的图形生成模型,用于端到端设计具有高药物相似性的化学和构象有效 3D 分子。然后,我们结合 L-Net 和 Monte Carlo 树搜索来执行基于结构的de novo药物设计任务。在 SARS-CoV-2 主要蛋白酶抑制剂设计的案例研究中,DeepLigBuilder 提出了一系列具有新化学结构、高预测亲和力和与已知抑制剂相似的结合特征的类药物化合物。当前版本的 L-Net 是针对来自 ChEMBL 的类药物化合物进行训练的,可以根据用户的需求轻松地将其扩展到具有所需特性的其他分子数据集,并应用于功能分子的生成。DeepLigBuilder 将深度生成模型与原子级相互作用评估相结合,为基于结构的从头药物设计和先导优化提供了最先进的模型。










































 京公网安备 11010802027423号
京公网安备 11010802027423号