当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Local energy decomposition analysis and molecular properties of encapsulated methane in fullerene (CH4@C60)
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2021-09-01 , DOI: 10.1039/d1cp02333k Aleksander Jaworski 1 , Niklas Hedin 1
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2021-09-01 , DOI: 10.1039/d1cp02333k Aleksander Jaworski 1 , Niklas Hedin 1
Affiliation
|
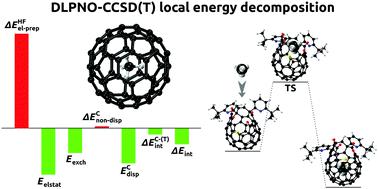
Methane has been successfully encapsulated within cages of C60 fullerene, which is an appropriate model system to study confinement effects. Its chemistry and physics are also relevant for theoretical model descriptions. Here we provide insights into intermolecular interactions and predicted spectroscopic responses of the CH4@C60 complex and compared them with results from other methods and with data from the literature. Local energy decomposition analysis (LED) within the domain-based local pair natural orbital coupled cluster singles, doubles, and perturbative triples (DLPNO-CCSD(T)) framework was used, and an efficient protocol for studies of endohedral complexes of fullerenes is proposed. This approach allowed us to assess energies in relation to electronic and geometric preparation, electrostatics, exchange, and London dispersion for the CH4@C60 endohedral complex. The calculated stabilization energy of CH4 inside the C60 fullerene was −13.5 kcal mol−1 and its magnitude was significantly larger than the latent heat of evaporation of CH4. Evaluation of vibrational frequencies and polarizabilities of the CH4@C60 complex revealed that the infrared (IR) and Raman bands of the endohedral CH4 were essentially “silent” due to the dielectric screening effect of C60, which acted as a molecular Faraday cage. Absorption spectra in the UV-vis domain and ionization potentials of C60 and CH4@C60 were predicted. They were almost identical. The calculated 1H/13C NMR shifts and spin–spin coupling constants were in very good agreement with experimental data. In addition, reference DLPNO-CCSD(T) interaction energies for complexes with noble gases (Ng@C60; Ng = He, Ne, Ar, Kr) were calculated. The values were compared with those derived from supramolecular MP2/SCS-MP2 calculations and estimates with London-type formulas by Pyykkö and coworkers [Phys. Chem. Chem. Phys., 2010, 12, 6187–6203], and with values derived from DFT-based symmetry-adapted perturbation theory (DFT-SAPT) by Hesselmann & Korona [Phys. Chem. Chem. Phys., 2011, 13, 732–743]. Selected points at the potential energy surface of the endohedral He2@C60 trimer were considered. In contrast to previous theoretical attempts with the DFT/MP2/SCS-MP2/DFT-SAPT methods, our calculations at the DLPNO-CCSD(T) level of theory predicted the He2@C60 trimer to be thermodynamically stable, which is in agreement with experimental observations.
中文翻译:

富勒烯(CH4@C60)中包封甲烷的局部能量分解分析和分子性质
甲烷已成功封装在 C 60富勒烯笼中,这是研究限制效应的合适模型系统。它的化学和物理也与理论模型描述相关。在这里,我们提供了对 CH 4 @C 60 的分子间相互作用和预测光谱响应的见解复杂,并将它们与其他方法的结果和文献中的数据进行比较。使用基于域的局部对自然轨道耦合簇单峰、双峰和微扰三元组 (DLPNO-CCSD(T)) 框架内的局部能量分解分析 (LED),提出了一种研究富勒烯内嵌配合物的有效方案. 这种方法使我们能够评估与 CH 4 @C 60内嵌配合物的电子和几何制备、静电学、交换和伦敦色散相关的能量。C 60富勒烯中 CH 4的计算稳定能为 -13.5 kcal mol -1且其大小明显大于CH 4的蒸发潜热。CH 4 @C 60复合物的振动频率和极化率的评估表明,由于 C 60的介电屏蔽效应,C 60充当分子法拉第,因此内嵌 CH 4的红外(IR)和拉曼带基本上是“沉默的”笼。预测了 C 60和 CH 4 @C 60的 UV-vis 域中的吸收光谱和电离电位。他们几乎一模一样。计算出的1 H/ 13C NMR 位移和自旋-自旋耦合常数与实验数据非常一致。此外,还计算了与稀有气体(Ng@C 60;Ng = He、Ne、Ar、Kr)的配合物的参考 DLPNO-CCSD(T) 相互作用能。将这些值与 Pyykkö 及其同事 [ Phys. 化学 化学 物理。, 2010, 12 , 6187–6203],以及 Hesselmann & Korona [ Phys. 化学 化学 物理。, 2011, 13 , 732–743]。在内嵌体 He 2 @C 60势能面上的选定点考虑了三聚体。与之前使用 DFT/MP2/SCS-MP2/DFT-SAPT 方法进行的理论尝试相比,我们在 DLPNO-CCSD(T) 理论水平的计算预测 He 2 @C 60三聚体是热力学稳定的,在与实验观察一致。
更新日期:2021-09-22
中文翻译:
富勒烯(CH4@C60)中包封甲烷的局部能量分解分析和分子性质
甲烷已成功封装在 C 60富勒烯笼中,这是研究限制效应的合适模型系统。它的化学和物理也与理论模型描述相关。在这里,我们提供了对 CH 4 @C 60 的分子间相互作用和预测光谱响应的见解复杂,并将它们与其他方法的结果和文献中的数据进行比较。使用基于域的局部对自然轨道耦合簇单峰、双峰和微扰三元组 (DLPNO-CCSD(T)) 框架内的局部能量分解分析 (LED),提出了一种研究富勒烯内嵌配合物的有效方案. 这种方法使我们能够评估与 CH 4 @C 60内嵌配合物的电子和几何制备、静电学、交换和伦敦色散相关的能量。C 60富勒烯中 CH 4的计算稳定能为 -13.5 kcal mol -1且其大小明显大于CH 4的蒸发潜热。CH 4 @C 60复合物的振动频率和极化率的评估表明,由于 C 60的介电屏蔽效应,C 60充当分子法拉第,因此内嵌 CH 4的红外(IR)和拉曼带基本上是“沉默的”笼。预测了 C 60和 CH 4 @C 60的 UV-vis 域中的吸收光谱和电离电位。他们几乎一模一样。计算出的1 H/ 13C NMR 位移和自旋-自旋耦合常数与实验数据非常一致。此外,还计算了与稀有气体(Ng@C 60;Ng = He、Ne、Ar、Kr)的配合物的参考 DLPNO-CCSD(T) 相互作用能。将这些值与 Pyykkö 及其同事 [ Phys. 化学 化学 物理。, 2010, 12 , 6187–6203],以及 Hesselmann & Korona [ Phys. 化学 化学 物理。, 2011, 13 , 732–743]。在内嵌体 He 2 @C 60势能面上的选定点考虑了三聚体。与之前使用 DFT/MP2/SCS-MP2/DFT-SAPT 方法进行的理论尝试相比,我们在 DLPNO-CCSD(T) 理论水平的计算预测 He 2 @C 60三聚体是热力学稳定的,在与实验观察一致。










































 京公网安备 11010802027423号
京公网安备 11010802027423号