Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Study on Chemical Bond Dissociation and the Removal of Oxygen-Containing Functional Groups of Low-Rank Coal during Hydrothermal Carbonization: DFT Calculations
ACS Omega ( IF 3.7 ) Pub Date : 2021-09-21 , DOI: 10.1021/acsomega.1c03866 Han Dang 1 , Guangwei Wang 1 , Chunmei Yu 1 , Xiaojun Ning 1 , Jianliang Zhang 1 , Nan Zhang 1 , Yi Gao 2 , Runsheng Xu 1 , Chuan Wang 3, 4
ACS Omega ( IF 3.7 ) Pub Date : 2021-09-21 , DOI: 10.1021/acsomega.1c03866 Han Dang 1 , Guangwei Wang 1 , Chunmei Yu 1 , Xiaojun Ning 1 , Jianliang Zhang 1 , Nan Zhang 1 , Yi Gao 2 , Runsheng Xu 1 , Chuan Wang 3, 4
Affiliation
|
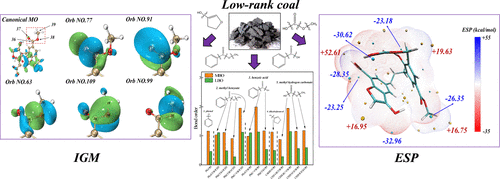
The molecular structure model of lignite was constructed, and the dissociation and removal mechanism of different C–O bonds and oxygen-containing functional groups was investigated using density functional theory (DFT) calculations. First, the bond order and bond dissociation enthalpy (BDE) were analyzed to predict the strength of different chemical bonds, and differences in the BDE and bond order were related to the difference in the fragment structure and electronic effects. The first group to break during hydrothermal carbonization (HTC) is the methyl of Ph(CO)O–CH3, followed by the C–O of CH3–OC(O)OH; the hydroxyl in Ph–OH is the most thermally stable group, followed by the hydroxyl in CH3OC(O)–OH. In addition, the orbital localization analysis has also been carried out. All three chemical bonds of Ph(CO)OCH3 show the characteristics of σ bond, while Ph(C═O)OCH3 and Ph(CO)–OCH3 with the Mayer bond order (MBO) greater than 1 also contains certain π bond characteristics. The lignite van der Waals (vdW) surface electrostatic potential (ESP) was constructed and visualized, and the results showed that the oxygen-containing functional groups mainly contributed to the area with a large absolute ESP. Finally, weak interactions between water molecules and lignite at different sites were described by independent gradient model (IGM) analysis. Models A, B, and E formed weak interactions with the hydrogen bond as the main force; model E showed the weakest hydrogen bond, while model C showed van der Waals interaction as the dominant force. In addition, some steric effect was also observed in model D.
中文翻译:

水热碳化过程中低阶煤化学键解离和含氧官能团去除的研究:DFT计算
构建了褐煤的分子结构模型,利用密度泛函理论(DFT)计算研究了不同C-O键和含氧官能团的解离和去除机理。首先,通过分析键序和键解离焓(BDE)来预测不同化学键的强度,BDE和键序的差异与碎片结构和电子效应的差异有关。在水热碳化 (HTC) 过程中,第一个断裂的基团是 Ph(CO)O–CH 3的甲基,其次是 CH 3 –OC(O)OH的 C–O ;Ph-OH 中的羟基是最热稳定的基团,其次是 CH 3 中的羟基OC(O)-OH。此外,还进行了轨道定位分析。Ph(CO)OCH 3 的三个化学键均表现出σ键的特性,而Ph(C=O)OCH 3和Ph(CO)-OCH 3具有大于 1 的 Mayer 键阶 (MBO) 也包含某些 π 键特性。构建并可视化褐煤范德华(vdW)表面静电势(ESP),结果表明含氧官能团主要贡献于绝对静电势大的区域。最后,通过独立梯度模型(IGM)分析描述了不同位置的水分子和褐煤之间的弱相互作用。模型A、B、E以氢键为主力形成弱相互作用;模型 E 显示出最弱的氢键,而模型 C 显示范德华相互作用为主导力。此外,在模型 D 中也观察到一些空间效应。
更新日期:2021-10-06
中文翻译:
水热碳化过程中低阶煤化学键解离和含氧官能团去除的研究:DFT计算
构建了褐煤的分子结构模型,利用密度泛函理论(DFT)计算研究了不同C-O键和含氧官能团的解离和去除机理。首先,通过分析键序和键解离焓(BDE)来预测不同化学键的强度,BDE和键序的差异与碎片结构和电子效应的差异有关。在水热碳化 (HTC) 过程中,第一个断裂的基团是 Ph(CO)O–CH 3的甲基,其次是 CH 3 –OC(O)OH的 C–O ;Ph-OH 中的羟基是最热稳定的基团,其次是 CH 3 中的羟基OC(O)-OH。此外,还进行了轨道定位分析。Ph(CO)OCH 3 的三个化学键均表现出σ键的特性,而Ph(C=O)OCH 3和Ph(CO)-OCH 3具有大于 1 的 Mayer 键阶 (MBO) 也包含某些 π 键特性。构建并可视化褐煤范德华(vdW)表面静电势(ESP),结果表明含氧官能团主要贡献于绝对静电势大的区域。最后,通过独立梯度模型(IGM)分析描述了不同位置的水分子和褐煤之间的弱相互作用。模型A、B、E以氢键为主力形成弱相互作用;模型 E 显示出最弱的氢键,而模型 C 显示范德华相互作用为主导力。此外,在模型 D 中也观察到一些空间效应。










































 京公网安备 11010802027423号
京公网安备 11010802027423号