当前位置:
X-MOL 学术
›
Comput. Struct. Biotechnol. J.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Getting to know each other: PPIMem, a novel approach for predicting transmembrane protein-protein complexes
Computational and Structural Biotechnology Journal ( IF 4.4 ) Pub Date : 2021-09-17 , DOI: 10.1016/j.csbj.2021.09.013 Georges Khazen 1 , Aram Gyulkhandanian 2 , Tina Issa 1 , Rachid C Maroun 2
Computational and Structural Biotechnology Journal ( IF 4.4 ) Pub Date : 2021-09-17 , DOI: 10.1016/j.csbj.2021.09.013 Georges Khazen 1 , Aram Gyulkhandanian 2 , Tina Issa 1 , Rachid C Maroun 2
Affiliation
|
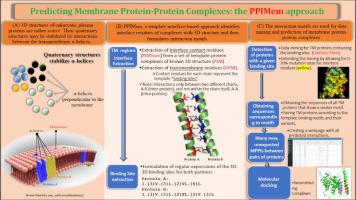
Because of their considerable number and diversity, membrane proteins and their macromolecular complexes represent the functional units of cells. Their quaternary structure may be stabilized by interactions between the α-helices of different proteins in the hydrophobic region of the cell membrane. Membrane proteins equally represent potential pharmacological targets par excellence for various diseases. Unfortunately, their experimental 3D structure and that of their complexes with other intramembrane protein partners are scarce due to technical difficulties. To overcome this key problem, we devised PPIMem, a computational approach for the specific prediction of higher-order structures of α-helical transmembrane proteins. The novel approach involves proper identification of the amino acid residues at the interface of molecular complexes with a 3D structure. The identified residues compose then nonlinear interaction motifs that are conveniently expressed as mathematical regular expressions. These are efficiently implemented for motif search in amino acid sequence databases, and for the accurate prediction of intramembrane protein-protein complexes. Our template interface-based approach predicted 21,544 binary complexes between 1,504 eukaryotic plasma membrane proteins across 39 species. We compare our predictions to experimental datasets of protein-protein interactions as a first validation method. The online database that results from the PPIMem algorithm with the annotated predicted interactions are implemented as a web server and can be accessed directly at .
中文翻译:

相互了解:PPIMem,一种预测跨膜蛋白-蛋白复合物的新方法
由于膜蛋白及其大分子复合物数量巨大且具有多样性,它们代表了细胞的功能单位。它们的四级结构可以通过细胞膜疏水区域中不同蛋白质的α螺旋之间的相互作用来稳定。膜蛋白同样代表了各种疾病的潜在药理学靶点。不幸的是,由于技术困难,它们的实验性 3D 结构以及它们与其他膜内蛋白伴侣的复合物很少。为了克服这个关键问题,我们设计了 PPIMem,一种用于特异性预测 α-螺旋跨膜蛋白高阶结构的计算方法。这种新方法涉及正确识别具有 3D 结构的分子复合物界面上的氨基酸残基。识别出的残基组成非线性相互作用基序,可以方便地表达为数学正则表达式。这些可以有效地实现氨基酸序列数据库中的基序搜索,以及膜内蛋白质-蛋白质复合物的准确预测。我们基于模板接口的方法预测了 39 个物种的 1,504 个真核细胞质膜蛋白之间的 21,544 个二元复合物。我们将我们的预测与蛋白质-蛋白质相互作用的实验数据集进行比较,作为第一种验证方法。由 PPIMem 算法生成的在线数据库以及带注释的预测交互被实现为 Web 服务器,并且可以直接在 访问。
更新日期:2021-09-17
中文翻译:
相互了解:PPIMem,一种预测跨膜蛋白-蛋白复合物的新方法
由于膜蛋白及其大分子复合物数量巨大且具有多样性,它们代表了细胞的功能单位。它们的四级结构可以通过细胞膜疏水区域中不同蛋白质的α螺旋之间的相互作用来稳定。膜蛋白同样代表了各种疾病的潜在药理学靶点。不幸的是,由于技术困难,它们的实验性 3D 结构以及它们与其他膜内蛋白伴侣的复合物很少。为了克服这个关键问题,我们设计了 PPIMem,一种用于特异性预测 α-螺旋跨膜蛋白高阶结构的计算方法。这种新方法涉及正确识别具有 3D 结构的分子复合物界面上的氨基酸残基。识别出的残基组成非线性相互作用基序,可以方便地表达为数学正则表达式。这些可以有效地实现氨基酸序列数据库中的基序搜索,以及膜内蛋白质-蛋白质复合物的准确预测。我们基于模板接口的方法预测了 39 个物种的 1,504 个真核细胞质膜蛋白之间的 21,544 个二元复合物。我们将我们的预测与蛋白质-蛋白质相互作用的实验数据集进行比较,作为第一种验证方法。由 PPIMem 算法生成的在线数据库以及带注释的预测交互被实现为 Web 服务器,并且可以直接在 访问。










































 京公网安备 11010802027423号
京公网安备 11010802027423号