Science Bulletin ( IF 18.8 ) Pub Date : 2021-09-17 , DOI: 10.1016/j.scib.2021.09.010 Qiangqiang Gu 1 , Linfeng Zhang 2 , Ji Feng 3
|
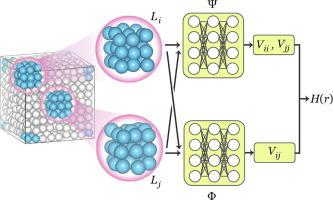
Despite their rich information content, electronic structure data amassed at high volumes in ab initio molecular dynamics simulations are generally under-utilized. We introduce a transferable high-fidelity neural network representation of such data in the form of tight-binding Hamiltonians for crystalline materials. This predictive representation of ab initio electronic structure, combined with machine-learning boosted molecular dynamics, enables efficient and accurate electronic evolution and sampling. When it is applied to a one-dimension charge-density wave material, carbyne, we are able to compute the spectral function and optical conductivity in the canonical ensemble. The spectral functions evaluated during soliton-antisoliton pair annihilation process reveal significant renormalization of low-energy edge modes due to retarded electron-lattice coupling beyond the Born–Oppenheimer limit. The availability of an efficient and reusable surrogate model for the electronic structure dynamical system will enable calculating many interesting physical properties, paving the way to previously inaccessible or challenging avenues in materials modeling.
中文翻译:
从头算分子动力学的电子结构的神经网络表示
尽管信息内容丰富,但在从头算分子动力学模拟中大量积累的电子结构数据通常未得到充分利用。我们以晶体材料的紧束缚哈密顿量的形式引入了此类数据的可转移高保真神经网络表示。这种从头算起的预测表示电子结构与机器学习增强的分子动力学相结合,可实现高效准确的电子进化和采样。当它应用于一维电荷密度波材料卡宾时,我们能够计算正则系综中的光谱函数和光导率。在孤子-反孤子对湮灭过程中评估的光谱函数揭示了由于延迟的电子-晶格耦合超过玻恩-奥本海默极限而导致的低能边缘模式的显着重整化。电子结构动力系统的高效且可重复使用的替代模型的可用性将使计算许多有趣的物理特性成为可能,为材料建模中以前无法接近或具有挑战性的途径铺平了道路。










































 京公网安备 11010802027423号
京公网安备 11010802027423号