当前位置:
X-MOL 学术
›
J. Phys. Chem. B
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Transferable Gaussian Attractive Potentials for Organic/Oxide Interfaces
The Journal of Physical Chemistry B ( IF 2.8 ) Pub Date : 2021-09-17 , DOI: 10.1021/acs.jpcb.1c05156 Jérôme Rey 1 , Sarah Blanck 1, 2 , Paul Clabaut 1 , Sophie Loehlé 2 , Stephan N Steinmann 1 , Carine Michel 1
The Journal of Physical Chemistry B ( IF 2.8 ) Pub Date : 2021-09-17 , DOI: 10.1021/acs.jpcb.1c05156 Jérôme Rey 1 , Sarah Blanck 1, 2 , Paul Clabaut 1 , Sophie Loehlé 2 , Stephan N Steinmann 1 , Carine Michel 1
Affiliation
|
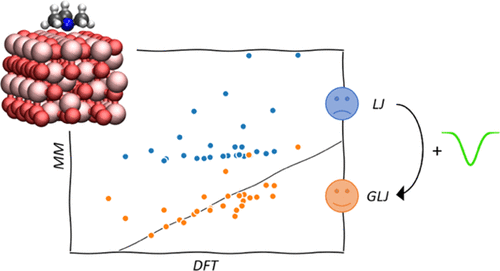
Organic/oxide interfaces play an important role in many areas of chemistry and in particular for lubrication and corrosion. Molecular dynamics simulations are the method of choice for providing complementary insight to experiments. However, the force fields used to simulate the interaction between molecules and oxide surfaces tend to capture only weak physisorption interactions, discarding the stabilizing Lewis acid/base interactions. We here propose a simple complement to the straightforward molecular mechanics description based on “out-of-the-box” Lennard-Jones potentials and electrostatic interactions: the addition of an attractive Gaussian potential between reactive sites of the surface and heteroatoms of adsorbed organic molecules, leading to the Gaussian Lennard-Jones (GLJ) potential. The interactions of four oxygenated and four amine molecules with the typical and widespread hematite and γ-alumina surfaces are investigated. The root mean square deviation (RMSD) for all probed molecules is only 5.7 kcal/mol, which corresponds to an error of 23% over hematite. On γ-alumina, the RMSD is 11.2 kcal/mol using a single parameter for all five chemically inequivalent surface aluminum atoms. Applying GLJ to the simulation of organic films on oxide surfaces demonstrates that the mobility of the surfactants is overestimated by the simplistic LJ potential, while GLJ and other qualitatively correct potentials show a strong structuration and slow dynamics of the surface films, as could be expected from the first-principles adsorption energies for model head groups.
中文翻译:

有机/氧化物界面的可转移高斯引力势
有机/氧化物界面在化学的许多领域中发挥着重要作用,特别是在润滑和腐蚀方面。分子动力学模拟是为实验提供补充见解的首选方法。然而,用于模拟分子和氧化物表面之间相互作用的力场往往只捕获微弱的物理吸附相互作用,而忽略了稳定的路易斯酸/碱相互作用。我们在这里提出了对基于“开箱即用”的 Lennard-Jones 势和静电相互作用的直接分子力学描述的简单补充:在表面的反应位点和吸附的有机分子的杂原子之间添加有吸引力的高斯势,导致高斯伦纳德-琼斯 (GLJ) 势。研究了四个含氧分子和四个胺分子与典型且广泛分布的赤铁矿和γ-氧化铝表面的相互作用。所有探测分子的均方根偏差 (RMSD) 仅为 5.7 kcal/mol,对应于赤铁矿 23% 的误差。在 γ-氧化铝上,对于所有五个化学不等价的表面铝原子,使用单一参数的 RMSD 为 11.2 kcal/mol。将 GLJ 应用于氧化物表面有机膜的模拟表明,表面活性剂的迁移率被简单化的 LJ 电位高估,而 GLJ 和其他定性正确的电位显示出表面膜的强结构和缓慢动力学,正如可以预期的那样模型头基团的第一性原理吸附能。
更新日期:2021-09-30
中文翻译:
有机/氧化物界面的可转移高斯引力势
有机/氧化物界面在化学的许多领域中发挥着重要作用,特别是在润滑和腐蚀方面。分子动力学模拟是为实验提供补充见解的首选方法。然而,用于模拟分子和氧化物表面之间相互作用的力场往往只捕获微弱的物理吸附相互作用,而忽略了稳定的路易斯酸/碱相互作用。我们在这里提出了对基于“开箱即用”的 Lennard-Jones 势和静电相互作用的直接分子力学描述的简单补充:在表面的反应位点和吸附的有机分子的杂原子之间添加有吸引力的高斯势,导致高斯伦纳德-琼斯 (GLJ) 势。研究了四个含氧分子和四个胺分子与典型且广泛分布的赤铁矿和γ-氧化铝表面的相互作用。所有探测分子的均方根偏差 (RMSD) 仅为 5.7 kcal/mol,对应于赤铁矿 23% 的误差。在 γ-氧化铝上,对于所有五个化学不等价的表面铝原子,使用单一参数的 RMSD 为 11.2 kcal/mol。将 GLJ 应用于氧化物表面有机膜的模拟表明,表面活性剂的迁移率被简单化的 LJ 电位高估,而 GLJ 和其他定性正确的电位显示出表面膜的强结构和缓慢动力学,正如可以预期的那样模型头基团的第一性原理吸附能。










































 京公网安备 11010802027423号
京公网安备 11010802027423号