当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Crossed-Beam and Theoretical Studies of the O(3P, 1D) + Benzene Reactions: Primary Products, Branching Fractions, and Role of Intersystem Crossing
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2021-09-17 , DOI: 10.1021/acs.jpca.1c06913 Gianmarco Vanuzzo 1 , Adriana Caracciolo 1 , Timothy K Minton 1 , Nadia Balucani 1 , Piergiorgio Casavecchia 1 , Carlo de Falco 2 , Alberto Baggioli 3 , Carlo Cavallotti 3
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2021-09-17 , DOI: 10.1021/acs.jpca.1c06913 Gianmarco Vanuzzo 1 , Adriana Caracciolo 1 , Timothy K Minton 1 , Nadia Balucani 1 , Piergiorgio Casavecchia 1 , Carlo de Falco 2 , Alberto Baggioli 3 , Carlo Cavallotti 3
Affiliation
|
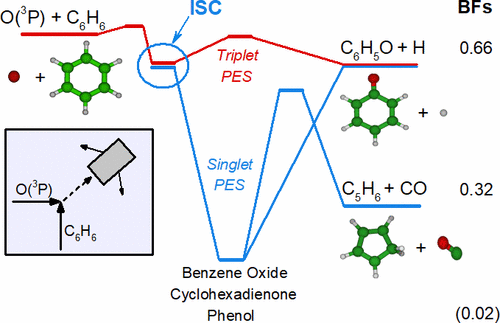
Reliable modeling of hydrocarbon oxidation relies critically on knowledge of the branching fractions (BFs) as a function of temperature (T) and pressure (p) for the products of the reaction of the hydrocarbon with atomic oxygen in its ground state, O(3P). During the past decade, we have performed in-depth investigations of the reactions of O(3P) with a variety of small unsaturated hydrocarbons using the crossed molecular beam (CMB) technique with universal mass spectrometric (MS) detection and time-of-flight (TOF) analysis, combined with synergistic theoretical calculations of the relevant potential energy surfaces (PESs) and statistical computations of product BFs, including intersystem crossing (ISC). This has allowed us to determine the primary products, their BFs, and extent of ISC to ultimately provide theoretical channel-specific rate constants as a function of T and p. In this work, we have extended this approach to the oxidation of one of the most important species involved in the combustion of aromatics: the benzene (C6H6) molecule. Despite extensive experimental and theoretical studies on the kinetics and dynamics of the O(3P) + C6H6 reaction, the relative importance of the C6H5O (phenoxy) + H open-shell products and of the spin-forbidden C5H6 (cyclopentadiene) + CO and phenol adduct closed-shell products are still open issues, which have hampered the development of reliable benzene combustion models. With the CMB technique, we have investigated the reaction dynamics of O(3P) + benzene at a collision energy (Ec) of 8.2 kcal/mol, focusing on the occurrence of the phenoxy + H and spin-forbidden C5H6 + CO and phenol channels in order to shed further light on the dynamics of this complex and important reaction, including the role of ISC. Concurrently, we have also investigated the reaction dynamics of O(1D) + benzene at the same Ec. Synergistic high-level electronic structure calculations of the underlying triplet/singlet PESs, including nonadiabatic couplings, have been performed to complement and assist the interpretation of the experimental results. Statistical (RRKM)/master equation (ME) computations of the product distribution and BFs on these PESs, with inclusion of ISC, have been performed and compared to experiment. In light of the reasonable agreement between the CMB experiment, literature kinetic experimental results, and theoretical predictions for the O(3P) + benzene reaction, the so-validated computational methodology has been used to predict (i) the BF between the C6H5O + H and C5H6 + CO channels as a function of collision energy and temperature (at 0.1 and 1 bar), showing that their increase progressively favors radical (phenoxy + H)-forming over molecule (C5H6 + CO and phenol stabilization)-forming channels, and (ii) channel-specific rate constants as a function of T and p, which are expected to be useful for improved combustion models.
中文翻译:

O(3P, 1D) + 苯反应的交叉束和理论研究:初级产物、支化分数和系统间交叉的作用
烃氧化的可靠建模关键依赖于支化分数 (BFs) 作为温度 ( T ) 和压力 ( p )的函数的知识,用于烃与基态原子氧反应的产物,O( 3 P )。在过去的十年中,我们使用具有通用性的交叉分子束 (CMB) 技术对 O( 3 P) 与各种小不饱和烃的反应进行了深入研究。质谱 (MS) 检测和飞行时间 (TOF) 分析,结合相关势能面 (PES) 的协同理论计算和产物 BF 的统计计算,包括系统间交叉 (ISC)。这使我们能够确定初级产品、它们的 BF 和 ISC 的范围,以最终提供作为T和p函数的理论通道特定速率常数。在这项工作中,我们将这种方法扩展到参与芳烃燃烧的最重要物质之一的氧化:苯 (C 6 H 6 ) 分子。尽管对 O( 3 P) + C 6的动力学和动力学进行了广泛的实验和理论研究H 6反应,C 6 H 5 O (苯氧基) + H 开壳产物和自旋禁止的 C 5 H 6 (环戊二烯) + CO 和苯酚加合物闭壳产物的相对重要性仍然是悬而未决的问题,这阻碍了可靠的苯燃烧模型的开发。使用 CMB 技术,我们研究了 O( 3 P) + 苯在8.2 kcal/mol碰撞能量 ( E c ) 下的反应动力学,重点研究了苯氧基 + H 和自旋禁止的 C 5 H 6 的发生+ CO 和苯酚通道,以进一步阐明这种复杂而重要的反应的动力学,包括 ISC 的作用。同时,我们还研究了 O( 1 D) + 苯在相同E c 下的反应动力学。已经进行了基础三重态/单重态 PES 的协同高级电子结构计算,包括非绝热耦合,以补充和帮助解释实验结果。这些 PES 上的产品分布和 BF 的统计 (RRKM)/主方程 (ME) 计算,包括 ISC,已经执行并与实验进行了比较。鉴于CMB实验、文献动力学实验结果和O(3 P) + 苯反应,如此验证的计算方法已用于预测 (i) C 6 H 5 O + H 和 C 5 H 6 + CO 通道之间的 BF作为碰撞能量和温度的函数(在0.1 和 1 bar),表明它们的增加逐渐有利于自由基(苯氧基 + H)形成而不是分子(C 5 H 6 + CO 和苯酚稳定化)形成通道,以及(ii)通道特定速率常数作为T和p,预计可用于改进的燃烧模型。
更新日期:2021-09-30
中文翻译:
O(3P, 1D) + 苯反应的交叉束和理论研究:初级产物、支化分数和系统间交叉的作用
烃氧化的可靠建模关键依赖于支化分数 (BFs) 作为温度 ( T ) 和压力 ( p )的函数的知识,用于烃与基态原子氧反应的产物,O( 3 P )。在过去的十年中,我们使用具有通用性的交叉分子束 (CMB) 技术对 O( 3 P) 与各种小不饱和烃的反应进行了深入研究。质谱 (MS) 检测和飞行时间 (TOF) 分析,结合相关势能面 (PES) 的协同理论计算和产物 BF 的统计计算,包括系统间交叉 (ISC)。这使我们能够确定初级产品、它们的 BF 和 ISC 的范围,以最终提供作为T和p函数的理论通道特定速率常数。在这项工作中,我们将这种方法扩展到参与芳烃燃烧的最重要物质之一的氧化:苯 (C 6 H 6 ) 分子。尽管对 O( 3 P) + C 6的动力学和动力学进行了广泛的实验和理论研究H 6反应,C 6 H 5 O (苯氧基) + H 开壳产物和自旋禁止的 C 5 H 6 (环戊二烯) + CO 和苯酚加合物闭壳产物的相对重要性仍然是悬而未决的问题,这阻碍了可靠的苯燃烧模型的开发。使用 CMB 技术,我们研究了 O( 3 P) + 苯在8.2 kcal/mol碰撞能量 ( E c ) 下的反应动力学,重点研究了苯氧基 + H 和自旋禁止的 C 5 H 6 的发生+ CO 和苯酚通道,以进一步阐明这种复杂而重要的反应的动力学,包括 ISC 的作用。同时,我们还研究了 O( 1 D) + 苯在相同E c 下的反应动力学。已经进行了基础三重态/单重态 PES 的协同高级电子结构计算,包括非绝热耦合,以补充和帮助解释实验结果。这些 PES 上的产品分布和 BF 的统计 (RRKM)/主方程 (ME) 计算,包括 ISC,已经执行并与实验进行了比较。鉴于CMB实验、文献动力学实验结果和O(3 P) + 苯反应,如此验证的计算方法已用于预测 (i) C 6 H 5 O + H 和 C 5 H 6 + CO 通道之间的 BF作为碰撞能量和温度的函数(在0.1 和 1 bar),表明它们的增加逐渐有利于自由基(苯氧基 + H)形成而不是分子(C 5 H 6 + CO 和苯酚稳定化)形成通道,以及(ii)通道特定速率常数作为T和p,预计可用于改进的燃烧模型。










































 京公网安备 11010802027423号
京公网安备 11010802027423号