当前位置:
X-MOL 学术
›
ChemistrySelect
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
New Hybrid (E)-4-((pyren-1-ylmethylene)amino)-N-(thiazol-2-yl)benzenesulfonamide as a Potential Drug Candidate: Spectroscopy, TD-DFT, NBO, FMO, and MEP Studies**
ChemistrySelect ( IF 1.9 ) Pub Date : 2021-09-15 , DOI: 10.1002/slct.202102602 Musa Erdoğan 1 , Goncagül Serdaroğlu 2
ChemistrySelect ( IF 1.9 ) Pub Date : 2021-09-15 , DOI: 10.1002/slct.202102602 Musa Erdoğan 1 , Goncagül Serdaroğlu 2
Affiliation
|
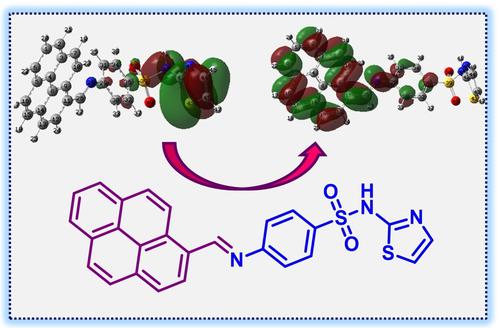
A novel pyrene-sulfathiazole-based potential drug candidate 3 was designed, successfully synthesized by a condensation reaction of pyrenecarboxaldehyde (1) with sulfathiazole (2) in good yield, and fully characterized by NMR, IR, UV-Vis, and HRMS spectroscopic techniques. The TD-DFT/B3LYP calculations displayed that the recorded peaks at 396, 377, 280, and 236 nm were due to the mainly n-π* and partially π-π* transitions that occurred on pyrene, azomethine, and sulfathiazole moieties. The NBO results disclosed that the electron delocalizations happened onto sulfathiazole, aromatic rings (pyrene and Ph-), and azomethine groups had the main responsibility of the lowering stabilization energy of compound 3. The NMR shifts were calculated by using the GIAO approaches in the DMSO phase and compared with the relevant data recorded. The thermodynamic and quantum chemical quantities were used for elucidating the physicochemical properties and reactivity behavior, in THF, DMSO, and water simulation environments. Accordingly, the calculated DM (D) and α (au) order of compound 3 were calculated in the order of vacuum<THF<DMSO<water, which indicated that the stability and thus reactivity of the compound strongly depended on the solvent environment. The FMO studies implied that the electron-donating possibility of compound 3 was dominant to the electron-accepting potency. In this work, the SMD version of IEFPCM was used in solvent media simulations.
中文翻译:

新型杂化 (E)-4-((pyren-1-ylmethylene)amino)-N-(thiazol-2-yl) 苯磺酰胺作为潜在候选药物:光谱、TD-DFT、NBO、FMO 和 MEP 研究**
设计了一种新型芘磺胺噻唑类潜在候选药物3,通过芘甲醛 ( 1 ) 与磺胺噻唑 ( 2 )的缩合反应以良好的收率成功合成,并通过 NMR、IR、UV-Vis 和 HRMS 光谱技术进行了充分表征. TD-DFT/B3LYP 计算表明,在 396、377、280 和 236 nm 处记录的峰主要是由于发生在芘、偶氮甲碱和磺胺噻唑部分上的 n-π* 跃迁和部分 π-π* 跃迁。NBO结果表明,电子离域发生在磺胺噻唑、芳环(芘和Ph-)和偶氮甲碱基团上,主要负责降低化合物3的稳定能. NMR 位移是通过在 DMSO 相中使用 GIAO 方法计算的,并与记录的相关数据进行比较。热力学和量子化学量用于阐明 THF、DMSO 和水模拟环境中的物理化学性质和反应行为。因此,计算化合物3的DM(D)和α(au)顺序是按照真空<THF<DMSO<水的顺序计算的,这表明化合物的稳定性和因此的反应性强烈依赖于溶剂环境。FMO 研究表明化合物 3 的给电子可能性对电子接受效力起主导作用。在这项工作中,IEFPCM 的 SMD 版本用于溶剂介质模拟。
更新日期:2021-09-15
中文翻译:
新型杂化 (E)-4-((pyren-1-ylmethylene)amino)-N-(thiazol-2-yl) 苯磺酰胺作为潜在候选药物:光谱、TD-DFT、NBO、FMO 和 MEP 研究**
设计了一种新型芘磺胺噻唑类潜在候选药物3,通过芘甲醛 ( 1 ) 与磺胺噻唑 ( 2 )的缩合反应以良好的收率成功合成,并通过 NMR、IR、UV-Vis 和 HRMS 光谱技术进行了充分表征. TD-DFT/B3LYP 计算表明,在 396、377、280 和 236 nm 处记录的峰主要是由于发生在芘、偶氮甲碱和磺胺噻唑部分上的 n-π* 跃迁和部分 π-π* 跃迁。NBO结果表明,电子离域发生在磺胺噻唑、芳环(芘和Ph-)和偶氮甲碱基团上,主要负责降低化合物3的稳定能. NMR 位移是通过在 DMSO 相中使用 GIAO 方法计算的,并与记录的相关数据进行比较。热力学和量子化学量用于阐明 THF、DMSO 和水模拟环境中的物理化学性质和反应行为。因此,计算化合物3的DM(D)和α(au)顺序是按照真空<THF<DMSO<水的顺序计算的,这表明化合物的稳定性和因此的反应性强烈依赖于溶剂环境。FMO 研究表明化合物 3 的给电子可能性对电子接受效力起主导作用。在这项工作中,IEFPCM 的 SMD 版本用于溶剂介质模拟。










































 京公网安备 11010802027423号
京公网安备 11010802027423号