当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
CHARMM-GUI Free Energy Calculator for Practical Ligand Binding Free Energy Simulations with AMBER
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2021-09-15 , DOI: 10.1021/acs.jcim.1c00747 Han Zhang 1 , Seonghoon Kim 2 , Timothy J Giese 3 , Tai-Sung Lee 3 , Jumin Lee 1 , Darrin M York 3 , Wonpil Im 1
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2021-09-15 , DOI: 10.1021/acs.jcim.1c00747 Han Zhang 1 , Seonghoon Kim 2 , Timothy J Giese 3 , Tai-Sung Lee 3 , Jumin Lee 1 , Darrin M York 3 , Wonpil Im 1
Affiliation
|
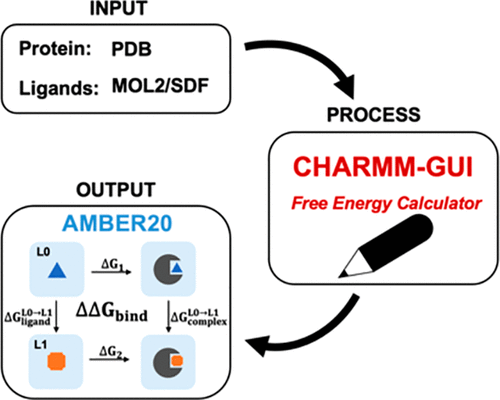
Alchemical free energy methods, such as free energy perturbation (FEP) and thermodynamic integration (TI), become increasingly popular and crucial for drug design and discovery. However, the system preparation of alchemical free energy simulation is an error-prone, time-consuming, and tedious process for a large number of ligands. To address this issue, we have recently presented CHARMM-GUI Free Energy Calculator that can provide input and postprocessing scripts for NAMD and GENESIS FEP molecular dynamics systems. In this work, we extended three submodules of Free Energy Calculator to work with the full suite of GPU-accelerated alchemical free energy methods and tools in AMBER, including input and postprocessing scripts. The BACE1 (β-secretase 1) benchmark set was used to validate the AMBER-TI simulation systems and scripts generated by Free Energy Calculator. The overall results of relatively large and diverse systems are almost equivalent with different protocols (unified and split) and with different timesteps (1, 2, and 4 fs), with R2 > 0.9. More importantly, the average free energy differences between two protocols are small and reliable with four independent runs, with a mean unsigned error (MUE) below 0.4 kcal/mol. Running at least four independent runs for each pair with AMBER20 (and FF19SB/GAFF2.1/OPC force fields), we obtained a MUE of 0.99 kcal/mol and root-mean-square error of 1.31 kcal/mol for 58 alchemical transformations in comparison with experimental data. In addition, a set of ligands for T4-lysozyme was used to further validate our free energy calculation protocol whose results are close to experimental data (within 1 kcal/mol). In summary, Free Energy Calculator provides a user-friendly web-based tool to generate the AMBER-TI system and input files for high-throughput binding free energy calculations with access to the full set of GPU-accelerated alchemical free energy, enhanced sampling, and analysis methods in AMBER.
中文翻译:

CHARMM-GUI 自由能计算器,用于使用 AMBER 进行实际配体结合自由能模拟
炼金术自由能方法,如自由能扰动 (FEP) 和热力学积分 (TI),在药物设计和发现中变得越来越流行和至关重要。然而,对于大量配体而言,炼金术自由能模拟的系统准备是一个容易出错、耗时且繁琐的过程。为了解决这个问题,我们最近推出了 CHARMM-GUI自由能计算器,它可以为 NAMD 和 GENESIS FEP 分子动力学系统提供输入和后处理脚本。在这项工作中,我们扩展了自由能计算器的三个子模块在 AMBER 中使用全套 GPU 加速炼金术自由能方法和工具,包括输入和后处理脚本。BACE1(β-分泌酶 1)基准集用于验证由自由能计算器生成的 AMBER-TI 仿真系统和脚本。相对较大且多样化的系统的总体结果几乎等同于不同的协议(统一和拆分)和不同的时间步长(1、2 和 4 fs),其中R 2> 0.9。更重要的是,两个协议之间的平均自由能差异小且可靠,具有四次独立运行,平均无符号误差 (MUE) 低于 0.4 kcal/mol。使用 AMBER20(和 FF19SB/GAFF2.1/OPC 力场)对每对运行至少四次独立运行,我们获得了 0.99 kcal/mol 的 MUE 和 1.31 kcal/mol 的均方根误差,用于 58 次炼金术转化与实验数据的比较。此外,一组 T4-溶菌酶的配体用于进一步验证我们的自由能计算方案,其结果接近实验数据(在 1 kcal/mol 以内)。总之,自由能计算器 提供了一个用户友好的基于 Web 的工具来生成 AMBER-TI 系统和输入文件,用于高通量结合自由能计算,并可以访问 AMBER 中的全套 GPU 加速炼金术自由能、增强采样和分析方法。
更新日期:2021-09-27
中文翻译:
CHARMM-GUI 自由能计算器,用于使用 AMBER 进行实际配体结合自由能模拟
炼金术自由能方法,如自由能扰动 (FEP) 和热力学积分 (TI),在药物设计和发现中变得越来越流行和至关重要。然而,对于大量配体而言,炼金术自由能模拟的系统准备是一个容易出错、耗时且繁琐的过程。为了解决这个问题,我们最近推出了 CHARMM-GUI自由能计算器,它可以为 NAMD 和 GENESIS FEP 分子动力学系统提供输入和后处理脚本。在这项工作中,我们扩展了自由能计算器的三个子模块在 AMBER 中使用全套 GPU 加速炼金术自由能方法和工具,包括输入和后处理脚本。BACE1(β-分泌酶 1)基准集用于验证由自由能计算器生成的 AMBER-TI 仿真系统和脚本。相对较大且多样化的系统的总体结果几乎等同于不同的协议(统一和拆分)和不同的时间步长(1、2 和 4 fs),其中R 2> 0.9。更重要的是,两个协议之间的平均自由能差异小且可靠,具有四次独立运行,平均无符号误差 (MUE) 低于 0.4 kcal/mol。使用 AMBER20(和 FF19SB/GAFF2.1/OPC 力场)对每对运行至少四次独立运行,我们获得了 0.99 kcal/mol 的 MUE 和 1.31 kcal/mol 的均方根误差,用于 58 次炼金术转化与实验数据的比较。此外,一组 T4-溶菌酶的配体用于进一步验证我们的自由能计算方案,其结果接近实验数据(在 1 kcal/mol 以内)。总之,自由能计算器 提供了一个用户友好的基于 Web 的工具来生成 AMBER-TI 系统和输入文件,用于高通量结合自由能计算,并可以访问 AMBER 中的全套 GPU 加速炼金术自由能、增强采样和分析方法。









































 京公网安备 11010802027423号
京公网安备 11010802027423号