当前位置:
X-MOL 学术
›
J. Phys. Chem. B
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Accurate and Transferable Reactive Molecular Dynamics Models from Constrained Density Functional Theory
The Journal of Physical Chemistry B ( IF 2.8 ) Pub Date : 2021-09-14 , DOI: 10.1021/acs.jpcb.1c05992 Chenghan Li 1 , Gregory A Voth 1
The Journal of Physical Chemistry B ( IF 2.8 ) Pub Date : 2021-09-14 , DOI: 10.1021/acs.jpcb.1c05992 Chenghan Li 1 , Gregory A Voth 1
Affiliation
|
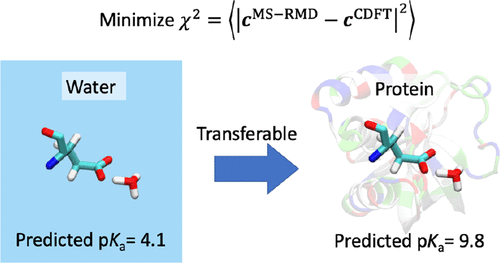
Chemical reactions constitute the central feature of many liquid, material, and biomolecular processes. Conventional molecular dynamics (MD) is inadequate for simulating chemical reactions given the fixed bonding topology of most force fields, while modeling chemical reactions using ab initio molecular dynamics is limited to shorter time and length scales given its high computational cost. As such, the multiscale reactive molecular dynamics method provides one promising alternative for simulating complex chemical systems at atomistic detail on a reactive potential energy surface. However, the parametrization of such models is a key barrier to their applicability and success. In this work, we present reactive MD models derived from constrained density functional theory that are both accurate and transferable. We illustrate the features of these models for proton dissociation reactions of amino acids in both aqueous and protein environments. Specifically, we present models for ionizable glutamate and lysine that predict accurate absolute pKa values in water as well as their significantly shifted pKa in staphylococcal nuclease (SNase) without any modification of the models. As one outcome of the new methodology, the simulations show that the deprotonation of ionizable residues in SNase can be closely coupled with side chain rotations, which is a concept likely generalizable to many other proteins. Furthermore, the present approach is not limited to only pKa prediction but can enable the fully atomistic simulation of many other reactive systems along with a determination of the key aspects of the reaction mechanisms.
中文翻译:

来自约束密度泛函理论的准确且可转移的反应分子动力学模型
化学反应构成了许多液体、材料和生物分子过程的核心特征。考虑到大多数力场的固定键拓扑,传统的分子动力学 (MD) 不足以模拟化学反应,而使用ab initio模拟化学反应鉴于其高计算成本,分子动力学仅限于更短的时间和长度尺度。因此,多尺度反应分子动力学方法为在反应势能表面上以原子细节模拟复杂化学系统提供了一种有前景的替代方法。然而,此类模型的参数化是其适用性和成功的主要障碍。在这项工作中,我们提出了从约束密度泛函理论派生的反应式 MD 模型,这些模型既准确又可转移。我们说明了这些模型在水性和蛋白质环境中氨基酸的质子解离反应的特征。具体来说,我们提出了可电离谷氨酸和赖氨酸的模型,可预测准确的绝对 p K a水中的值以及它们在葡萄球菌核酸酶 (SNase) 中的p K a显着变化,而无需对模型进行任何修改。作为新方法的一个结果,模拟表明 SNase 中可电离残基的去质子化可以与侧链旋转密切相关,这是一个可能适用于许多其他蛋白质的概念。此外,本方法不仅限于 p K a预测,而且可以使许多其他反应系统的完全原子模拟以及反应机制的关键方面的确定成为可能。
更新日期:2021-09-23
中文翻译:
来自约束密度泛函理论的准确且可转移的反应分子动力学模型
化学反应构成了许多液体、材料和生物分子过程的核心特征。考虑到大多数力场的固定键拓扑,传统的分子动力学 (MD) 不足以模拟化学反应,而使用ab initio模拟化学反应鉴于其高计算成本,分子动力学仅限于更短的时间和长度尺度。因此,多尺度反应分子动力学方法为在反应势能表面上以原子细节模拟复杂化学系统提供了一种有前景的替代方法。然而,此类模型的参数化是其适用性和成功的主要障碍。在这项工作中,我们提出了从约束密度泛函理论派生的反应式 MD 模型,这些模型既准确又可转移。我们说明了这些模型在水性和蛋白质环境中氨基酸的质子解离反应的特征。具体来说,我们提出了可电离谷氨酸和赖氨酸的模型,可预测准确的绝对 p K a水中的值以及它们在葡萄球菌核酸酶 (SNase) 中的p K a显着变化,而无需对模型进行任何修改。作为新方法的一个结果,模拟表明 SNase 中可电离残基的去质子化可以与侧链旋转密切相关,这是一个可能适用于许多其他蛋白质的概念。此外,本方法不仅限于 p K a预测,而且可以使许多其他反应系统的完全原子模拟以及反应机制的关键方面的确定成为可能。








































 京公网安备 11010802027423号
京公网安备 11010802027423号