当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Corrections in the CHARMM36 Parametrization of Chloride Interactions with Proteins, Lipids, and Alkali Cations, and Extension to Other Halide Anions
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2021-09-13 , DOI: 10.1021/acs.jctc.1c00550 Esam A Orabi 1 , Tuǧba N Öztürk 1, 2 , Nathan Bernhardt 1 , José D Faraldo-Gómez 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2021-09-13 , DOI: 10.1021/acs.jctc.1c00550 Esam A Orabi 1 , Tuǧba N Öztürk 1, 2 , Nathan Bernhardt 1 , José D Faraldo-Gómez 1
Affiliation
|
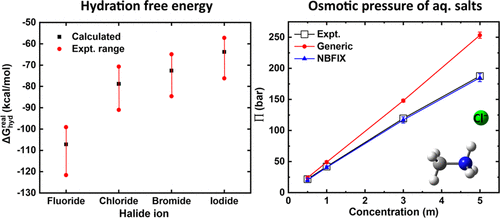
The nonpolarizable CHARMM force field is one of the most widely used energy functions for all-atom biomolecular simulations. Chloride is the only halide ion included in the latest version, CHARMM36m, and is used widely in simulation studies, often as an electrolyte ion but also as the biological substrate of transport proteins and enzymes. Here, we find that existing parameters systematically underestimate the interaction of Cl– with proteins and lipids. Accordingly, when examined in solution, little to no Cl–association can be observed with most components of the protein, including backbone, polar side chains and aromatic rings. The strength of the interaction with cationic side chains and with alkali ions is also incongruent with experimental measurements, specifically osmotic coefficients of concentrated solutions. Consistent with these findings, a 4-μs trajectory of the Cl–-specific transport protein CLC-ec1 shows irreversible Cl– dissociation from the so-called Scen binding site, even in a 150 mM NaCl buffer. To correct for these deficiencies, we formulate a series of pair-specific Lennard-Jones parameters that override those resulting from the conventional Lorentz–Berthelot combination rules. These parameters, referred to as NBFIX, are systematically calibrated against available experimental data as well as ab initio geometry optimizations and energy evaluations, for a wide set of binary and ternary Cl– complexes with protein and lipid analogs and alkali cations. Analogously, we also formulate parameter sets for the other three biological halide ions, namely, fluoride, bromide, and iodide. The resulting parameters are used to calculate the potential of mean force defining the interaction of each anion and each of the protein and lipid analogues in bulk water, revealing association free energies in the range of −0.3 to −3.3 kcal/mol, with the F– complexes being the least stable. The NBFIX corrections also preserve the Cl– occupancy of CLC-ec1 in a second 4-μs trajectory. We posit that these optimized molecular-mechanics models provide a more realistic foundation for all-atom simulation studies of processes entailing changes in hydration, recognition, or transport of halide anions.
中文翻译:

更正 CHARMM36 氯离子与蛋白质、脂质和碱阳离子相互作用的参数化,以及扩展到其他卤化物阴离子
不可极化 CHARMM 力场是全原子生物分子模拟中使用最广泛的能量函数之一。氯离子是最新版本 CHARMM36m 中唯一包含的卤离子,广泛用于模拟研究,通常用作电解质离子,也用作转运蛋白和酶的生物底物。在这里,我们发现,现有的系统参数低估氯的互动-与蛋白质和脂类。因此,当在溶液中检查时,几乎没有 Cl –可以观察到与蛋白质的大多数成分的结合,包括主链、极性侧链和芳香环。与阳离子侧链和碱离子相互作用的强度也与实验测量不一致,特别是浓溶液的渗透系数。与这些发现一致,Cl -特异性转运蛋白 CLC-ec1的 4 μs 轨迹显示出不可逆的 Cl -从所谓的 S cen解离结合位点,即使在 150 mM NaCl 缓冲液中。为了纠正这些缺陷,我们制定了一系列特定对的 Lennard-Jones 参数,这些参数覆盖了由传统 Lorentz-Berthelot 组合规则产生的参数。这些参数(称为 NBFIX)针对可用的实验数据以及从头算几何优化和能量评估进行系统校准,适用于广泛的二元和三元 Cl –与蛋白质和脂质类似物以及碱金属阳离子的复合物。类似地,我们还制定了其他三种生物卤化物离子的参数集,即氟化物、溴化物和碘化物。得到的参数用于计算定义散装水中每种阴离子和每种蛋白质和脂质类似物相互作用的平均力势,揭示了在 -0.3 到 -3.3 kcal/mol 范围内的缔合自由能,与 F –配合物是最不稳定的。该NBFIX修正也保持氯- CLC-EC1的占用在第二个4微秒的轨迹。我们认为,这些优化的分子力学模型为全原子模拟研究需要改变卤化物阴离子的水合、识别或传输的过程提供了更现实的基础。
更新日期:2021-10-12
中文翻译:
更正 CHARMM36 氯离子与蛋白质、脂质和碱阳离子相互作用的参数化,以及扩展到其他卤化物阴离子
不可极化 CHARMM 力场是全原子生物分子模拟中使用最广泛的能量函数之一。氯离子是最新版本 CHARMM36m 中唯一包含的卤离子,广泛用于模拟研究,通常用作电解质离子,也用作转运蛋白和酶的生物底物。在这里,我们发现,现有的系统参数低估氯的互动-与蛋白质和脂类。因此,当在溶液中检查时,几乎没有 Cl –可以观察到与蛋白质的大多数成分的结合,包括主链、极性侧链和芳香环。与阳离子侧链和碱离子相互作用的强度也与实验测量不一致,特别是浓溶液的渗透系数。与这些发现一致,Cl -特异性转运蛋白 CLC-ec1的 4 μs 轨迹显示出不可逆的 Cl -从所谓的 S cen解离结合位点,即使在 150 mM NaCl 缓冲液中。为了纠正这些缺陷,我们制定了一系列特定对的 Lennard-Jones 参数,这些参数覆盖了由传统 Lorentz-Berthelot 组合规则产生的参数。这些参数(称为 NBFIX)针对可用的实验数据以及从头算几何优化和能量评估进行系统校准,适用于广泛的二元和三元 Cl –与蛋白质和脂质类似物以及碱金属阳离子的复合物。类似地,我们还制定了其他三种生物卤化物离子的参数集,即氟化物、溴化物和碘化物。得到的参数用于计算定义散装水中每种阴离子和每种蛋白质和脂质类似物相互作用的平均力势,揭示了在 -0.3 到 -3.3 kcal/mol 范围内的缔合自由能,与 F –配合物是最不稳定的。该NBFIX修正也保持氯- CLC-EC1的占用在第二个4微秒的轨迹。我们认为,这些优化的分子力学模型为全原子模拟研究需要改变卤化物阴离子的水合、识别或传输的过程提供了更现实的基础。










































 京公网安备 11010802027423号
京公网安备 11010802027423号