当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Expanded Ensemble Methods Can be Used to Accurately Predict Protein-Ligand Relative Binding Free Energies
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2021-09-13 , DOI: 10.1021/acs.jctc.1c00513 Si Zhang 1 , David F Hahn 2 , Michael R Shirts 3 , Vincent A Voelz 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2021-09-13 , DOI: 10.1021/acs.jctc.1c00513 Si Zhang 1 , David F Hahn 2 , Michael R Shirts 3 , Vincent A Voelz 1
Affiliation
|
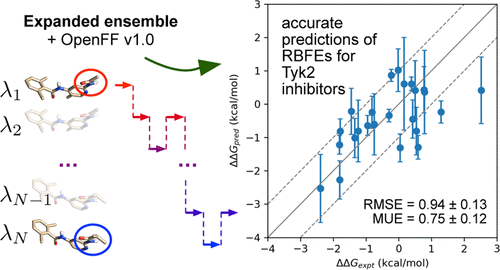
Alchemical free energy methods have become indispensable in computational drug discovery for their ability to calculate highly accurate estimates of protein-ligand affinities. Expanded ensemble (EE) methods, which involve single simulations visiting all of the alchemical intermediates, have some key advantages for alchemical free energy calculation. However, there have been relatively few examples published in the literature of using expanded ensemble simulations for free energies of protein-ligand binding. In this paper, as a test of expanded ensemble methods, we compute relative binding free energies using the Open Force Field Initiative force field (codename “Parsley”) for 24 pairs of Tyk2 inhibitors derived from a congeneric series of 16 compounds. The EE predictions agree well with the experimental values (root-mean-square error (RMSE) of 0.94 ± 0.13 kcal mol–1 and mean unsigned error (MUE) of 0.75 ± 0.12 kcal mol–1). We find that while increasing the number of alchemical intermediates can improve the phase space overlap, faster convergence can be obtained with fewer intermediates, as long as acceptance rates are sufficient. We also find that convergence can be improved using more aggressive updating of biases, and that estimates can be improved by performing multiple independent EE calculations. This work demonstrates that EE is a viable option for alchemical free energy calculation. We discuss the implications of these findings for rational drug design, as well as future directions for improvement.
中文翻译:

扩展集成方法可用于准确预测蛋白质-配体相对结合自由能
炼金术自由能方法已成为计算药物发现中不可或缺的方法,因为它们能够计算蛋白质-配体亲和力的高度准确估计。扩展系综 (EE) 方法涉及访问所有炼金术中间体的单一模拟,在炼金术自由能计算方面具有一些关键优势。然而,文献中发表的使用扩展集合模拟来获得蛋白质-配体结合的自由能的例子相对较少。在本文中,作为扩展集成方法的测试,我们使用 Open Force Field Initiative 力场(代号“Parsley”)计算来自 16 种同源化合物系列的 24 对 Tyk2 抑制剂的相对结合自由能。EE 预测与实验值(均方根误差 (RMSE) 为 0 非常吻合。–1和 0.75 ± 0.12 kcal mol –1的平均无符号误差 (MUE )。我们发现,虽然增加炼金中间体的数量可以改善相空间重叠,但只要接受率足够,可以用更少的中间体获得更快的收敛。我们还发现,可以通过更积极地更新偏差来提高收敛性,并且可以通过执行多个独立的 EE 计算来改进估计。这项工作表明 EE 是计算炼金术自由能的可行选择。我们讨论了这些发现对合理药物设计的影响,以及未来的改进方向。
更新日期:2021-10-12
中文翻译:
扩展集成方法可用于准确预测蛋白质-配体相对结合自由能
炼金术自由能方法已成为计算药物发现中不可或缺的方法,因为它们能够计算蛋白质-配体亲和力的高度准确估计。扩展系综 (EE) 方法涉及访问所有炼金术中间体的单一模拟,在炼金术自由能计算方面具有一些关键优势。然而,文献中发表的使用扩展集合模拟来获得蛋白质-配体结合的自由能的例子相对较少。在本文中,作为扩展集成方法的测试,我们使用 Open Force Field Initiative 力场(代号“Parsley”)计算来自 16 种同源化合物系列的 24 对 Tyk2 抑制剂的相对结合自由能。EE 预测与实验值(均方根误差 (RMSE) 为 0 非常吻合。–1和 0.75 ± 0.12 kcal mol –1的平均无符号误差 (MUE )。我们发现,虽然增加炼金中间体的数量可以改善相空间重叠,但只要接受率足够,可以用更少的中间体获得更快的收敛。我们还发现,可以通过更积极地更新偏差来提高收敛性,并且可以通过执行多个独立的 EE 计算来改进估计。这项工作表明 EE 是计算炼金术自由能的可行选择。我们讨论了这些发现对合理药物设计的影响,以及未来的改进方向。










































 京公网安备 11010802027423号
京公网安备 11010802027423号