当前位置:
X-MOL 学术
›
Cryst. Growth Des.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Automated In Silico Energy Mapping of Facet-Specific Interparticle Interactions
Crystal Growth & Design ( IF 3.2 ) Pub Date : 2021-09-10 , DOI: 10.1021/acs.cgd.1c00674 Alexandru A. Moldovan 1, 2 , Radoslav Y. Penchev 3 , Robert B. Hammond 4 , Jakub P. Janowiak 1 , Thomas E. Hardcastle 1, 2 , Andrew G. P. Maloney 2 , Simon D. A. Connell 5
Crystal Growth & Design ( IF 3.2 ) Pub Date : 2021-09-10 , DOI: 10.1021/acs.cgd.1c00674 Alexandru A. Moldovan 1, 2 , Radoslav Y. Penchev 3 , Robert B. Hammond 4 , Jakub P. Janowiak 1 , Thomas E. Hardcastle 1, 2 , Andrew G. P. Maloney 2 , Simon D. A. Connell 5
Affiliation
|
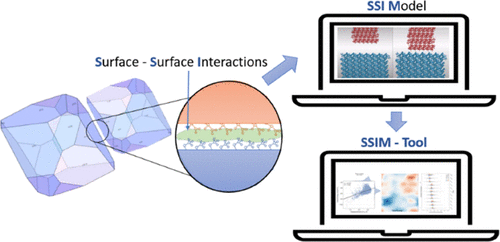
Particle–particle interactions impact the processability and performance of drug products. Faceted particulates exhibit distinct surface chemistries that affect their adhesion, causing downstream processing challenges such as poor flow, punch sticking, and compaction. Currently, there is a lack of tools to assist formulators in predicting these challenges based on particle properties. Here, we present a methodology for navigating the energy landscape of interparticle interactions. We used molecular mechanics to calculate the interactions between slabs of molecules representing distinct facets. The workflow enables a rapid assessment of the total energy landscape between interacting particles, providing insight into the effects of different surface chemistries and molecular topologies. Previously, the strongest interaction (lowest energy) was used to calculate the propensity to adhere, but we demonstrate that this does not always predict an accurate description of the likely surface interactions. We chose paracetamol to demonstrate the application of this methodology. The most cohesive facets were (101) and (10-1). Comparing surface interactions between particles allows a ranking of the most energetically compatible surfaces. The significance of this ranking and understanding how surface chemistry can impact interparticle interactions is a step toward assisting formulation decisions and improvements in product performance.
中文翻译:

特定面的粒子间相互作用的自动计算机内能量映射
颗粒间相互作用会影响药品的可加工性和性能。多面颗粒表现出不同的表面化学性质,会影响其附着力,从而导致下游加工挑战,例如流动性差、冲头粘连和压实。目前,缺乏帮助配方设计师根据颗粒特性预测这些挑战的工具。在这里,我们提出了一种导航粒子间相互作用的能量景观的方法。我们使用分子力学来计算代表不同方面的分子板之间的相互作用。该工作流程可以快速评估相互作用粒子之间的总能量图谱,从而深入了解不同表面化学和分子拓扑结构的影响。之前,最强相互作用(最低能量)用于计算粘附倾向,但我们证明这并不总是预测可能的表面相互作用的准确描述。我们选择扑热息痛来演示这种方法的应用。最具凝聚力的方面是 (101) 和 (10-1)。比较粒子之间的表面相互作用可以对能量最兼容的表面进行排序。该排名的重要性和了解表面化学如何影响颗粒间相互作用是帮助制定配方决策和提高产品性能的一步。我们选择扑热息痛来演示这种方法的应用。最具凝聚力的方面是 (101) 和 (10-1)。比较粒子之间的表面相互作用可以对能量最兼容的表面进行排序。该排名的重要性以及了解表面化学如何影响颗粒间相互作用是帮助制定配方决策和提高产品性能的一步。我们选择扑热息痛来演示这种方法的应用。最具凝聚力的方面是 (101) 和 (10-1)。比较粒子之间的表面相互作用可以对能量最兼容的表面进行排序。该排名的重要性以及了解表面化学如何影响颗粒间相互作用是帮助制定配方决策和提高产品性能的一步。
更新日期:2021-10-06
中文翻译:
特定面的粒子间相互作用的自动计算机内能量映射
颗粒间相互作用会影响药品的可加工性和性能。多面颗粒表现出不同的表面化学性质,会影响其附着力,从而导致下游加工挑战,例如流动性差、冲头粘连和压实。目前,缺乏帮助配方设计师根据颗粒特性预测这些挑战的工具。在这里,我们提出了一种导航粒子间相互作用的能量景观的方法。我们使用分子力学来计算代表不同方面的分子板之间的相互作用。该工作流程可以快速评估相互作用粒子之间的总能量图谱,从而深入了解不同表面化学和分子拓扑结构的影响。之前,最强相互作用(最低能量)用于计算粘附倾向,但我们证明这并不总是预测可能的表面相互作用的准确描述。我们选择扑热息痛来演示这种方法的应用。最具凝聚力的方面是 (101) 和 (10-1)。比较粒子之间的表面相互作用可以对能量最兼容的表面进行排序。该排名的重要性和了解表面化学如何影响颗粒间相互作用是帮助制定配方决策和提高产品性能的一步。我们选择扑热息痛来演示这种方法的应用。最具凝聚力的方面是 (101) 和 (10-1)。比较粒子之间的表面相互作用可以对能量最兼容的表面进行排序。该排名的重要性以及了解表面化学如何影响颗粒间相互作用是帮助制定配方决策和提高产品性能的一步。我们选择扑热息痛来演示这种方法的应用。最具凝聚力的方面是 (101) 和 (10-1)。比较粒子之间的表面相互作用可以对能量最兼容的表面进行排序。该排名的重要性以及了解表面化学如何影响颗粒间相互作用是帮助制定配方决策和提高产品性能的一步。










































 京公网安备 11010802027423号
京公网安备 11010802027423号