当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Tracking the Transition from Pericyclic to Pseudopericyclic Reaction Mechanisms Using Multicenter Electron Delocalization Analysis: The [1,3] Sigmatropic Rearrangement
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2021-09-11 , DOI: 10.1021/acs.jpca.1c06620 Álvaro Pérez-Barcia 1 , Ángeles Peña-Gallego 1 , Marcos Mandado 1
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2021-09-11 , DOI: 10.1021/acs.jpca.1c06620 Álvaro Pérez-Barcia 1 , Ángeles Peña-Gallego 1 , Marcos Mandado 1
Affiliation
|
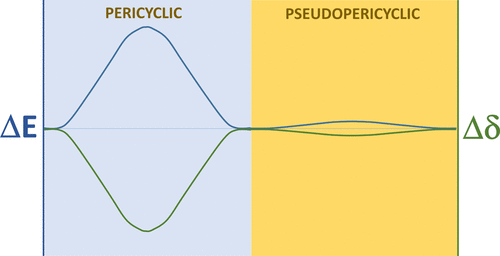
Herein, the power of multicenter electron delocalization analysis to elucidate the intricacies of concerted reaction mechanisms is brought to light by tracking the transition of [1,3] sigmatropic rearrangements from the high-barrier pericyclic mechanism in 1-butene to the barrierless pseudopericyclic mechanism in 1,2-diamino-1-nitrosooxyethane. This transition has been progressively achieved by substituting the migrating group, changing the donor and acceptor atoms, and functionalizing the alkene unit with weak and strong electron-donating and electron-withdrawing groups. Fourteen [1,3] sigmatropic reactions with electronic energy barriers ranging from 1 to 89 kcal/mol have been investigated. A very good correlation has been found between the barrier and the four-center electron delocalization at the transition state, the latter calculated for the atoms involved in the four-centered ring adduct formed along the reaction path. Surprisingly, the barrier has been found to be independent of the bond strength between the migrating group and the donor atom so that only the changes induced in the multicenter bonding control the kinetics of the reaction. Additional insights into the effect of atom substitution and group functionalization have also been extracted from the analysis of the multicenter electron delocalization profiles along the reaction path and qualitatively supported by the topological analysis of the electron density.
中文翻译:

使用多中心电子离域分析跟踪从周环到伪周环反应机制的转变:[1,3] Sigmatropic 重排
在此,通过跟踪 [1,3] σ 重排从 1-丁烯中的高势垒周环机制到无势垒伪周环机制的转变,揭示了多中心电子离域分析的力量,以阐明协同反应机制的复杂性。 1,2-二氨基-1-亚硝基氧乙烷。这种转变是通过取代迁移基团、改变供体和受体原子以及用弱和强给电子和吸电子基团官能化烯烃单元来逐步实现的。已经研究了十四个 [1,3] σ 反应,其电子能垒范围为 1 到 89 kcal/mol。在过渡态的势垒和四中心电子离域之间发现了非常好的相关性,后者计算了沿反应路径形成的四中心环加合物中涉及的原子。令人惊讶的是,已经发现势垒与迁移基团和供体原子之间的键强度无关,因此只有在多中心键合中引起的变化才能控制反应动力学。还从沿反应路径的多中心电子离域分布分析中提取了对原子取代和基团官能化影响的其他见解,并得到了电子密度拓扑分析的定性支持。已经发现屏障与迁移基团和供体原子之间的键强度无关,因此只有在多中心键合中引起的变化才能控制反应动力学。还从沿反应路径的多中心电子离域分布分析中提取了对原子取代和基团官能化影响的其他见解,并得到了电子密度拓扑分析的定性支持。已经发现屏障与迁移基团和供体原子之间的键强度无关,因此只有在多中心键合中引起的变化才能控制反应动力学。还从沿反应路径的多中心电子离域分布分析中提取了对原子取代和基团官能化影响的其他见解,并得到了电子密度拓扑分析的定性支持。
更新日期:2021-09-23
中文翻译:
使用多中心电子离域分析跟踪从周环到伪周环反应机制的转变:[1,3] Sigmatropic 重排
在此,通过跟踪 [1,3] σ 重排从 1-丁烯中的高势垒周环机制到无势垒伪周环机制的转变,揭示了多中心电子离域分析的力量,以阐明协同反应机制的复杂性。 1,2-二氨基-1-亚硝基氧乙烷。这种转变是通过取代迁移基团、改变供体和受体原子以及用弱和强给电子和吸电子基团官能化烯烃单元来逐步实现的。已经研究了十四个 [1,3] σ 反应,其电子能垒范围为 1 到 89 kcal/mol。在过渡态的势垒和四中心电子离域之间发现了非常好的相关性,后者计算了沿反应路径形成的四中心环加合物中涉及的原子。令人惊讶的是,已经发现势垒与迁移基团和供体原子之间的键强度无关,因此只有在多中心键合中引起的变化才能控制反应动力学。还从沿反应路径的多中心电子离域分布分析中提取了对原子取代和基团官能化影响的其他见解,并得到了电子密度拓扑分析的定性支持。已经发现屏障与迁移基团和供体原子之间的键强度无关,因此只有在多中心键合中引起的变化才能控制反应动力学。还从沿反应路径的多中心电子离域分布分析中提取了对原子取代和基团官能化影响的其他见解,并得到了电子密度拓扑分析的定性支持。已经发现屏障与迁移基团和供体原子之间的键强度无关,因此只有在多中心键合中引起的变化才能控制反应动力学。还从沿反应路径的多中心电子离域分布分析中提取了对原子取代和基团官能化影响的其他见解,并得到了电子密度拓扑分析的定性支持。










































 京公网安备 11010802027423号
京公网安备 11010802027423号