当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Modeling Kinetics and Thermodynamics of Guest Encapsulation into the [M4L6]12– Supramolecular Organometallic Cage
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2021-09-10 , DOI: 10.1021/acs.jcim.1c00348 Gantulga Norjmaa 1 , Pietro Vidossich 2 , Jean-Didier Maréchal 1 , Gregori Ujaque 1
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2021-09-10 , DOI: 10.1021/acs.jcim.1c00348 Gantulga Norjmaa 1 , Pietro Vidossich 2 , Jean-Didier Maréchal 1 , Gregori Ujaque 1
Affiliation
|
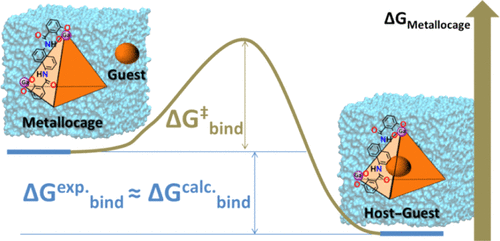
The encapsulation of molecular guests into supramolecular hosts is a complex molecular recognition process in which the guest displaces the solvent from the host cavity, while the host deforms to let the guest in. An atomistic description of the association would provide valuable insights on the physicochemical properties that guide it. This understanding may be used to design novel host assemblies with improved properties (i.e., affinities) toward a given class of guests. Molecular simulations may be conveniently used to model the association processes. It is thus of interest to establish efficient protocols to trace the encapsulation process and to predict the associated magnitudes ΔGbind and ΔGbind⧧. Here, we report the calculation of the Gibbs energy barrier and Gibbs binding energy by means of explicit solvent molecular simulations for the [Ga4L6]12– metallocage encapsulating a series of cationic molecules. The ΔGbind⧧ for encapsulation was estimated by means of umbrella sampling simulations. The steps involved were identified, including ion-pair formation and naphthalene rotation (from L ligands of the metallocage) during the guest’s entrance. The ΔGbind values were computed using the attach–pull–release method. The results reveal the sensitivity of the estimates on the force field parameters, in particular on atomic charges, showing that higher accuracy is obtained when charges are derived from implicit solvent quantum chemical calculations. Correlation analysis identified some indicators for the binding affinity trends. All computed magnitudes are in very good agreement with experimental observations. This work provides, on one side, a benchmarked way to computationally model a highly charged metallocage encapsulation process. This includes a nonstandard parameterization and charge derivation procedure. On the other hand, it gives specific mechanistic information on the binding processes of [Ga4L6]12– at the molecular level where key motions are depicted. Taken together, the study provides an interesting option for the future design of metal–organic cages.
中文翻译:

将客体封装到 [M4L6]12– 超分子有机金属笼中的动力学和热力学建模
将分子客体封装到超分子主体中是一个复杂的分子识别过程,其中客体从主体腔中置换溶剂,而主体变形以让客体进入。对缔合的原子描述将为物理化学性质提供有价值的见解那引导它。这种理解可用于针对给定类别的客人设计具有改进特性(即,亲和性)的新型宿主组件。分子模拟可方便地用于对关联过程建模。因此,建立有效的协议来追踪封装过程并预测相关的幅度 Δ G bind和 Δ G bind ⧧ 是很有意义的。. 在这里,我们报告了通过显式溶剂分子模拟对封装一系列阳离子分子的 [Ga 4 L 6 ] 12-金属位点的吉布斯能垒和吉布斯结合能的计算。用于封装的Δ G bind ⧧是通过伞形采样模拟来估计的。确定了所涉及的步骤,包括在客人进入期间形成离子对和萘旋转(来自金属位点的 L 配体)。该Δ ģ绑定使用附加-拉动-释放方法计算值。结果揭示了力场参数估计的敏感性,特别是原子电荷,表明当电荷从隐式溶剂量子化学计算中获得时,可以获得更高的精度。相关性分析确定了结合亲和力趋势的一些指标。所有计算出的星等都与实验观察非常吻合。一方面,这项工作提供了一种基准方法,可以对高电荷金属位点封装过程进行计算建模。这包括非标准的参数化和电荷推导程序。另一方面,它给出了[Ga 4 L 6 ] 12–结合过程的具体机理信息。在描述关键运动的分子水平上。总之,该研究为金属-有机笼的未来设计提供了一个有趣的选择。
更新日期:2021-09-27
中文翻译:
将客体封装到 [M4L6]12– 超分子有机金属笼中的动力学和热力学建模
将分子客体封装到超分子主体中是一个复杂的分子识别过程,其中客体从主体腔中置换溶剂,而主体变形以让客体进入。对缔合的原子描述将为物理化学性质提供有价值的见解那引导它。这种理解可用于针对给定类别的客人设计具有改进特性(即,亲和性)的新型宿主组件。分子模拟可方便地用于对关联过程建模。因此,建立有效的协议来追踪封装过程并预测相关的幅度 Δ G bind和 Δ G bind ⧧ 是很有意义的。. 在这里,我们报告了通过显式溶剂分子模拟对封装一系列阳离子分子的 [Ga 4 L 6 ] 12-金属位点的吉布斯能垒和吉布斯结合能的计算。用于封装的Δ G bind ⧧是通过伞形采样模拟来估计的。确定了所涉及的步骤,包括在客人进入期间形成离子对和萘旋转(来自金属位点的 L 配体)。该Δ ģ绑定使用附加-拉动-释放方法计算值。结果揭示了力场参数估计的敏感性,特别是原子电荷,表明当电荷从隐式溶剂量子化学计算中获得时,可以获得更高的精度。相关性分析确定了结合亲和力趋势的一些指标。所有计算出的星等都与实验观察非常吻合。一方面,这项工作提供了一种基准方法,可以对高电荷金属位点封装过程进行计算建模。这包括非标准的参数化和电荷推导程序。另一方面,它给出了[Ga 4 L 6 ] 12–结合过程的具体机理信息。在描述关键运动的分子水平上。总之,该研究为金属-有机笼的未来设计提供了一个有趣的选择。










































 京公网安备 11010802027423号
京公网安备 11010802027423号