当前位置:
X-MOL 学术
›
Biopolymers
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
A sequence-based method for predicting extant fold switchers that undergo α-helix ↔ β-strand transitions
Biopolymers ( IF 3.2 ) Pub Date : 2021-09-09 , DOI: 10.1002/bip.23471 Soumya Mishra 1, 2 , Loren L Looger 2 , Lauren L Porter 1, 3
Biopolymers ( IF 3.2 ) Pub Date : 2021-09-09 , DOI: 10.1002/bip.23471 Soumya Mishra 1, 2 , Loren L Looger 2 , Lauren L Porter 1, 3
Affiliation
|
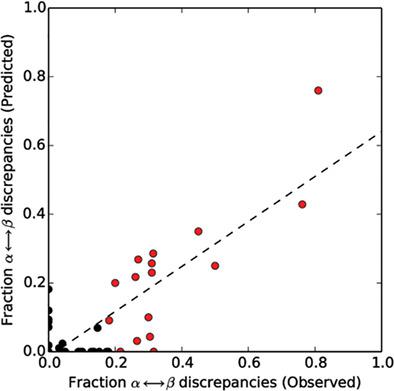
Extant fold-switching proteins remodel their secondary structures and change their functions in response to cellular stimuli, regulating biological processes and affecting human health. Despite their biological importance, these proteins remain understudied. Predictive methods are needed to expedite the process of discovering and characterizing more of these shapeshifting proteins. Most previous approaches require a solved structure or all-atom simulations, greatly constraining their use. Here, we propose a high-throughput sequence-based method for predicting extant fold switchers that transition from α-helix in one conformation to β-strand in the other. This method leverages two previous observations: (a) α-helix ↔ β-strand prediction discrepancies from JPred4 are a robust predictor of fold switching, and (b) the fold-switching regions (FSRs) of some extant fold switchers have different secondary structure propensities when expressed by themselves (isolated FSRs) than when expressed within the context of their parent protein (contextualized FSRs). Combining these two observations, we ran JPred4 on 99-fold-switching proteins and found strong correspondence between predicted and experimentally observed α-helix ↔ β-strand discrepancies. To test the overall robustness of this finding, we randomly selected regions of proteins not expected to switch folds (single-fold proteins) and found significantly fewer predicted α-helix ↔ β-strand discrepancies. Combining these discrepancies with the overall percentage of predicted secondary structure, we developed a classifier to identify extant fold switchers (Matthews correlation coefficient of .71). Although this classifier had a high false-negative rate (7/17), its false-positive rate was very low (2/136), suggesting that it can be used to predict a subset of extant fold switchers from a multitude of available genomic sequences.
中文翻译:

一种基于序列的方法,用于预测经历 α 螺旋 ↔ β 链转变的现存折叠转换蛋白
现存的折叠转换蛋白重塑其二级结构并改变其功能以响应细胞刺激,调节生物过程并影响人类健康。尽管它们具有重要的生物学意义,但这些蛋白质仍然未被充分研究。需要预测方法来加快发现和表征更多这些变形蛋白的过程。以前的大多数方法都需要解决结构或全原子模拟,这极大地限制了它们的使用。在这里,我们提出了一种基于高通量序列的方法,用于预测现有的折叠转换蛋白,这些折叠转换蛋白从一种构象中的α螺旋转变为另一种构象中的β链。该方法利用了之前的两个观察结果:(a) JPred4 的 α 螺旋 ↔ β 链预测差异是折叠转换的稳健预测因子,(b) 一些现有折叠转换蛋白的折叠转换区域 (FSR) 具有不同的二级结构与在其亲本蛋白上下文中表达(上下文化 FSR)相比,它们自身表达(孤立的 FSR)时的倾向。结合这两个观察结果,我们对 99 倍转换蛋白运行 JPred4,发现预测的 α 螺旋 ↔ β 链差异与实验观察到的差异之间存在很强的对应性。为了测试这一发现的整体稳健性,我们随机选择了预计不会转换折叠的蛋白质区域(单折叠蛋白质),并发现预测的 α 螺旋↔ β 链差异显着减少。将这些差异与预测二级结构的总体百分比相结合,我们开发了一个分类器来识别现有的折叠转换蛋白(Matthews 相关系数为 0.71)。 尽管该分类器具有较高的假阴性率 (7/17),但其假阳性率非常低 (2/136),这表明它可用于从大量可用的基因组中预测现有折叠转换子的子集序列。
更新日期:2021-10-25
中文翻译:
一种基于序列的方法,用于预测经历 α 螺旋 ↔ β 链转变的现存折叠转换蛋白
现存的折叠转换蛋白重塑其二级结构并改变其功能以响应细胞刺激,调节生物过程并影响人类健康。尽管它们具有重要的生物学意义,但这些蛋白质仍然未被充分研究。需要预测方法来加快发现和表征更多这些变形蛋白的过程。以前的大多数方法都需要解决结构或全原子模拟,这极大地限制了它们的使用。在这里,我们提出了一种基于高通量序列的方法,用于预测现有的折叠转换蛋白,这些折叠转换蛋白从一种构象中的α螺旋转变为另一种构象中的β链。该方法利用了之前的两个观察结果:(a) JPred4 的 α 螺旋 ↔ β 链预测差异是折叠转换的稳健预测因子,(b) 一些现有折叠转换蛋白的折叠转换区域 (FSR) 具有不同的二级结构与在其亲本蛋白上下文中表达(上下文化 FSR)相比,它们自身表达(孤立的 FSR)时的倾向。结合这两个观察结果,我们对 99 倍转换蛋白运行 JPred4,发现预测的 α 螺旋 ↔ β 链差异与实验观察到的差异之间存在很强的对应性。为了测试这一发现的整体稳健性,我们随机选择了预计不会转换折叠的蛋白质区域(单折叠蛋白质),并发现预测的 α 螺旋↔ β 链差异显着减少。将这些差异与预测二级结构的总体百分比相结合,我们开发了一个分类器来识别现有的折叠转换蛋白(Matthews 相关系数为 0.71)。 尽管该分类器具有较高的假阴性率 (7/17),但其假阳性率非常低 (2/136),这表明它可用于从大量可用的基因组中预测现有折叠转换子的子集序列。










































 京公网安备 11010802027423号
京公网安备 11010802027423号