当前位置:
X-MOL 学术
›
Aging Cell
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Increased mitochondrial calcium uptake and concomitant mitochondrial activity by presenilin loss promotes mTORC1 signaling to drive neurodegeneration
Aging Cell ( IF 8.0 ) Pub Date : 2021-09-09 , DOI: 10.1111/acel.13472 Kerry C Ryan 1 , Zahra Ashkavand 1 , Shaarika Sarasija 1 , Jocelyn T Laboy 1 , Rohan Samarakoon 1 , Kenneth R Norman 1
Aging Cell ( IF 8.0 ) Pub Date : 2021-09-09 , DOI: 10.1111/acel.13472 Kerry C Ryan 1 , Zahra Ashkavand 1 , Shaarika Sarasija 1 , Jocelyn T Laboy 1 , Rohan Samarakoon 1 , Kenneth R Norman 1
Affiliation
|
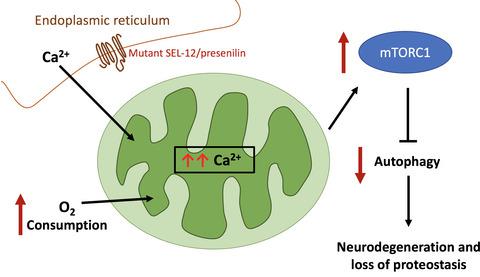
Metabolic dysfunction and protein aggregation are common characteristics that occur in age-related neurodegenerative disease. However, the mechanisms underlying these abnormalities remain poorly understood. We have found that mutations in the gene encoding presenilin in Caenorhabditis elegans, sel-12, results in elevated mitochondrial activity that drives oxidative stress and neuronal dysfunction. Mutations in the human presenilin genes are the primary cause of familial Alzheimer's disease. Here, we demonstrate that loss of SEL-12/presenilin results in the hyperactivation of the mTORC1 pathway. This hyperactivation is caused by elevated mitochondrial calcium influx and, likely, the associated increase in mitochondrial activity. Reducing mTORC1 activity improves proteostasis defects and neurodegenerative phenotypes associated with loss of SEL-12 function. Consistent with high mTORC1 activity, we find that SEL-12 loss reduces autophagosome formation, and this reduction is prevented by limiting mitochondrial calcium uptake. Moreover, the improvements of proteostasis and neuronal defects in sel-12 mutants due to mTORC1 inhibition require the induction of autophagy. These results indicate that mTORC1 hyperactivation exacerbates the defects in proteostasis and neuronal function in sel-12 mutants and demonstrate a critical role of presenilin in promoting neuronal health.
中文翻译:

早老素损失导致线粒体钙摄取增加和伴随的线粒体活性促进 mTORC1 信号传导以驱动神经退行性变
代谢功能障碍和蛋白质聚集是与年龄相关的神经退行性疾病的常见特征。然而,这些异常背后的机制仍然知之甚少。我们发现在秀丽隐杆线虫中编码早老素的基因sel - 12发生突变。,导致线粒体活性升高,从而导致氧化应激和神经元功能障碍。人类早老素基因的突变是家族性阿尔茨海默病的主要原因。在这里,我们证明 SEL-12/早老素的缺失会导致 mTORC1 通路的过度激活。这种过度活化是由线粒体钙内流升高引起的,并且很可能是线粒体活性的相关增加。降低 mTORC1 活性可改善与 SEL-12 功能丧失相关的蛋白质稳态缺陷和神经退行性表型。与 mTORC1 的高活性一致,我们发现 SEL-12 的丢失会减少自噬体的形成,而这种减少是通过限制线粒体钙摄取来防止的。此外,蛋白质稳态和神经元缺陷的改善 由于 mTORC1 抑制,12 个突变体需要诱导自噬。这些结果表明 mTORC1 过度激活加剧了sel - 12 突变体中蛋白质稳态和神经元功能的缺陷,并证明了早老素在促进神经元健康中的关键作用。
更新日期:2021-10-17
中文翻译:
早老素损失导致线粒体钙摄取增加和伴随的线粒体活性促进 mTORC1 信号传导以驱动神经退行性变
代谢功能障碍和蛋白质聚集是与年龄相关的神经退行性疾病的常见特征。然而,这些异常背后的机制仍然知之甚少。我们发现在秀丽隐杆线虫中编码早老素的基因sel - 12发生突变。,导致线粒体活性升高,从而导致氧化应激和神经元功能障碍。人类早老素基因的突变是家族性阿尔茨海默病的主要原因。在这里,我们证明 SEL-12/早老素的缺失会导致 mTORC1 通路的过度激活。这种过度活化是由线粒体钙内流升高引起的,并且很可能是线粒体活性的相关增加。降低 mTORC1 活性可改善与 SEL-12 功能丧失相关的蛋白质稳态缺陷和神经退行性表型。与 mTORC1 的高活性一致,我们发现 SEL-12 的丢失会减少自噬体的形成,而这种减少是通过限制线粒体钙摄取来防止的。此外,蛋白质稳态和神经元缺陷的改善 由于 mTORC1 抑制,12 个突变体需要诱导自噬。这些结果表明 mTORC1 过度激活加剧了sel - 12 突变体中蛋白质稳态和神经元功能的缺陷,并证明了早老素在促进神经元健康中的关键作用。








































 京公网安备 11010802027423号
京公网安备 11010802027423号