当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
CG2AT2: an Enhanced Fragment-Based Approach for Serial Multi-scale Molecular Dynamics Simulations
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2021-09-07 , DOI: 10.1021/acs.jctc.1c00295 Owen N Vickery 1 , Phillip J Stansfeld 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2021-09-07 , DOI: 10.1021/acs.jctc.1c00295 Owen N Vickery 1 , Phillip J Stansfeld 1
Affiliation
|
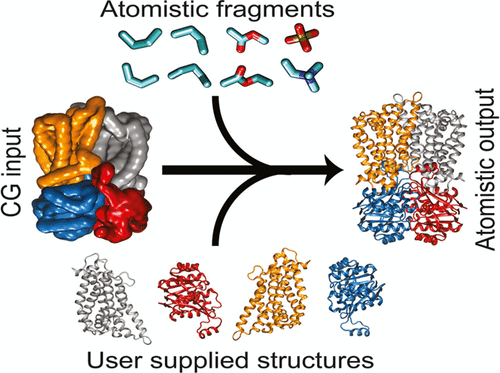
Coarse-grained molecular dynamics provides a means for simulating the assembly and interactions of macromolecular complexes at a reduced level of representation, thereby allowing both longer timescale and larger sized simulations. Here, we describe an enhanced fragment-based protocol for converting macromolecular complexes from coarse-grained to atomistic resolution, for further refinement and analysis. While the focus is upon systems that comprise an integral membrane protein embedded in a phospholipid bilayer, the technique is also suitable for membrane-anchored and soluble protein/nucleotide complexes. Overall, this provides a method for generating an accurate and well-equilibrated atomic-level description of a macromolecular complex. The approach is evaluated using a diverse test set of 11 system configurations of varying size and complexity. Simulations are assessed in terms of protein stereochemistry, conformational drift, lipid/protein interactions, and lipid dynamics.
中文翻译:

CG2AT2:一种增强的基于片段的串行多尺度分子动力学模拟方法
粗粒度分子动力学提供了一种以降低的表示水平模拟大分子复合物的组装和相互作用的方法,从而允许更长的时间尺度和更大的模拟。在这里,我们描述了一种增强的基于片段的协议,用于将大分子复合物从粗粒度分辨率转换为原子分辨率,以便进一步细化和分析。虽然重点是包含嵌入磷脂双层中的完整膜蛋白的系统,但该技术也适用于膜锚定和可溶性蛋白质/核苷酸复合物。总体而言,这提供了一种生成大分子复合物准确且平衡良好的原子级描述的方法。该方法使用包含 11 个不同大小和复杂性的系统配置的多样化测试集进行评估。
更新日期:2021-10-12
中文翻译:
CG2AT2:一种增强的基于片段的串行多尺度分子动力学模拟方法
粗粒度分子动力学提供了一种以降低的表示水平模拟大分子复合物的组装和相互作用的方法,从而允许更长的时间尺度和更大的模拟。在这里,我们描述了一种增强的基于片段的协议,用于将大分子复合物从粗粒度分辨率转换为原子分辨率,以便进一步细化和分析。虽然重点是包含嵌入磷脂双层中的完整膜蛋白的系统,但该技术也适用于膜锚定和可溶性蛋白质/核苷酸复合物。总体而言,这提供了一种生成大分子复合物准确且平衡良好的原子级描述的方法。该方法使用包含 11 个不同大小和复杂性的系统配置的多样化测试集进行评估。










































 京公网安备 11010802027423号
京公网安备 11010802027423号