当前位置:
X-MOL 学术
›
Biochemistry
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
A Streamlined Data Analysis Pipeline for the Identification of Sites of Citrullination
Biochemistry ( IF 2.9 ) Pub Date : 2021-09-07 , DOI: 10.1021/acs.biochem.1c00369 Aaron J Maurais 1 , Ari J Salinger 1, 2 , Micaela Tobin 2 , Scott A Shaffer 2, 3 , Eranthie Weerapana 1 , Paul R Thompson 2
Biochemistry ( IF 2.9 ) Pub Date : 2021-09-07 , DOI: 10.1021/acs.biochem.1c00369 Aaron J Maurais 1 , Ari J Salinger 1, 2 , Micaela Tobin 2 , Scott A Shaffer 2, 3 , Eranthie Weerapana 1 , Paul R Thompson 2
Affiliation
|
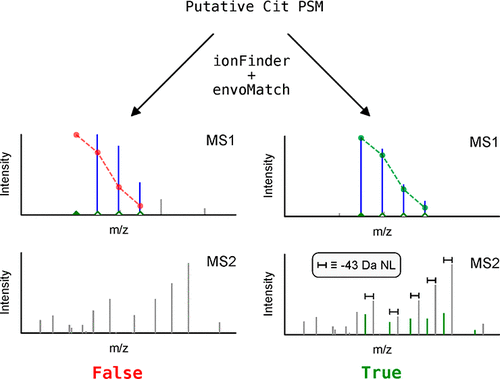
Citrullination is an enzyme-catalyzed post-translational modification (PTM) that is essential for a host of biological processes, including gene regulation, programmed cell death, and organ development. While this PTM is required for normal cellular functions, aberrant citrullination is a hallmark of autoimmune disorders as well as cancer. Although aberrant citrullination is linked to human pathology, the exact role of citrullination in disease remains poorly characterized, in part because of the challenges associated with identifying the specific arginine residues that are citrullinated. Tandem mass spectrometry is the most precise method for uncovering sites of citrullination; however, due to the small mass shift (+0.984 Da) that results from citrullination, current database search algorithms commonly misannotate spectra, leading to a high number of false-positive assignments. To address this challenge, we developed an automated workflow to rigorously and rapidly mine proteomic data to unambiguously identify the sites of citrullination from complex peptide mixtures. The crux of this streamlined workflow is the ionFinder software program, which classifies citrullination sites with high confidence on the basis of the presence of diagnostic fragment ions. These diagnostic ions include the neutral loss of isocyanic acid, which is a dissociative event that is unique to citrulline residues. Using the ionFinder program, we have mapped the sites of autocitrullination on purified protein arginine deiminases (PAD1–4) and mapped the global citrullinome in a PAD2-overexpressing cell line. The ionFinder algorithm is a highly versatile, user-friendly, and open-source program that is agnostic to the type of instrument and mode of fragmentation that are used.
中文翻译:

用于识别瓜氨酸位点的简化数据分析流程
瓜氨酸化是一种酶催化的翻译后修饰 (PTM),它对许多生物过程至关重要,包括基因调控、程序性细胞死亡和器官发育。虽然这种 PTM 是正常细胞功能所必需的,但异常瓜氨酸化是自身免疫性疾病和癌症的标志。尽管异常瓜氨酸化与人类病理学有关,但瓜氨酸化在疾病中的确切作用仍然缺乏表征,部分原因是与识别瓜氨酸化的特定精氨酸残基相关的挑战。串联质谱法是发现瓜氨酸化位点的最精确方法;然而,由于瓜氨酸化导致的质量偏移很小(+0.984 Da),当前的数据库搜索算法通常会错误注释光谱,导致大量的假阳性分配。为了应对这一挑战,我们开发了一种自动化工作流程,以严格快速地挖掘蛋白质组数据,以明确识别复杂肽混合物中的瓜氨酸化位点。这种简化工作流程的关键是 ionFinder 软件程序,它根据诊断碎片离子的存在高度可信地对瓜氨酸化位点进行分类。这些诊断离子包括异氰酸的中性损失,这是瓜氨酸残基特有的解离事件。使用 ionFinder 程序,我们绘制了纯化蛋白精氨酸脱亚胺酶 (PAD1-4) 上的自瓜氨酸化位点,并绘制了 PAD2 过表达细胞系中的全局瓜氨酸组。ionFinder 算法是一种高度通用、用户友好、
更新日期:2021-09-28
中文翻译:
用于识别瓜氨酸位点的简化数据分析流程
瓜氨酸化是一种酶催化的翻译后修饰 (PTM),它对许多生物过程至关重要,包括基因调控、程序性细胞死亡和器官发育。虽然这种 PTM 是正常细胞功能所必需的,但异常瓜氨酸化是自身免疫性疾病和癌症的标志。尽管异常瓜氨酸化与人类病理学有关,但瓜氨酸化在疾病中的确切作用仍然缺乏表征,部分原因是与识别瓜氨酸化的特定精氨酸残基相关的挑战。串联质谱法是发现瓜氨酸化位点的最精确方法;然而,由于瓜氨酸化导致的质量偏移很小(+0.984 Da),当前的数据库搜索算法通常会错误注释光谱,导致大量的假阳性分配。为了应对这一挑战,我们开发了一种自动化工作流程,以严格快速地挖掘蛋白质组数据,以明确识别复杂肽混合物中的瓜氨酸化位点。这种简化工作流程的关键是 ionFinder 软件程序,它根据诊断碎片离子的存在高度可信地对瓜氨酸化位点进行分类。这些诊断离子包括异氰酸的中性损失,这是瓜氨酸残基特有的解离事件。使用 ionFinder 程序,我们绘制了纯化蛋白精氨酸脱亚胺酶 (PAD1-4) 上的自瓜氨酸化位点,并绘制了 PAD2 过表达细胞系中的全局瓜氨酸组。ionFinder 算法是一种高度通用、用户友好、










































 京公网安备 11010802027423号
京公网安备 11010802027423号