当前位置:
X-MOL 学术
›
Chem. Biodivers.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Unravelling the Selectivity of 6,7-Dimethyl Quinoxaline Analogs for Kinase Inhibition: An Insight towards the Development of Alzheimer's Therapeutics
Chemistry & Biodiversity ( IF 2.9 ) Pub Date : 2021-09-05 , DOI: 10.1002/cbdv.202100364 Arvind Kumar Jain 1 , Arindam Gupta 2 , C Karthikeyan 3 , Piyush Trivedi 1, 4 , Anita Dutt Konar 1, 5, 6
Chemistry & Biodiversity ( IF 2.9 ) Pub Date : 2021-09-05 , DOI: 10.1002/cbdv.202100364 Arvind Kumar Jain 1 , Arindam Gupta 2 , C Karthikeyan 3 , Piyush Trivedi 1, 4 , Anita Dutt Konar 1, 5, 6
Affiliation
|
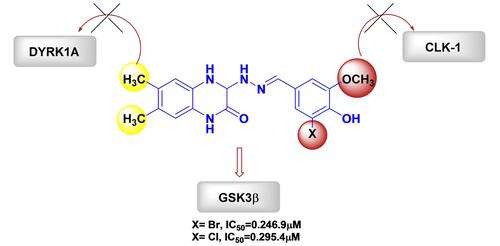
Untangling the most selective kinase inhibitors via pharmacological intervention remains one of the challenging affairs to date. In accordance to this drift, herein we describe the design and synthesis of a set of new heterocyclic analogs consisting of 6,7-dimethyl Quinoxaline, appended to a connector, employing Schiff base strategy (Compounds I–IX). The compounds were characterized by various spectroscopic techniques and the kinase inhibition assay were performed on few prime members of the CMGC family namely the GSK3β, DYRK1A and CLK1 receptors, respectively, that have been known to be directly involved in hyperphosphorylation of Tau. Interestingly the biological evaluation results revealed that Compounds IV and V, with bromo/chloro functionalities in the aromatic core were advantaged of being highly selective towards the target GSK3β over others. To strengthen our analysis, we adopted molecular modelling studies, where compounds IV/V were redocked in the same grid 4AFJ, as that of the reference ligand, 5-aryl-4-carboxamide-1,3-oxazole. Surprisingly, our investigation underpinned that for both the compounds IV/V, a primary H-bonding existed between the designed molecules (IV/V) and Val 135 residue in the receptor GSK3β, in line with the reference ligand. We attribute this interaction to instigate potency in the compounds. Indeed the other non-covalent interaction, between the derivative's aromatic nucleus and Arg 141/Thr 138 in the receptor GSK3β, might have been responsible for enhancing the selectivity in the targets. Overall, we feel that the present work depicts a logical demonstration towards fine tuning the efficacy of the inhibitors through systematic adjustment of electron density at appropriate positions in the aromatic ring be it the main quinoxaline or the other aromatic nucleus. Thus this pathway offers a convenient strategy for the development of efficient therapeutics for diversified neurodegenerative diseases like that of Alzheimer's.
中文翻译:

揭示 6,7-二甲基喹喔啉类似物对激酶抑制的选择性:对阿尔茨海默病治疗学发展的见解
通过药物干预解开最具选择性的激酶抑制剂仍然是迄今为止具有挑战性的事务之一。根据这种漂移,本文描述了一组新的杂环类似物的设计和合成,该类似物由附加到连接器的 6,7-二甲基喹喔啉组成,采用席夫碱策略(化合物I – IX)。通过各种光谱技术对化合物进行表征,并对 CMGC 家族的少数主要成员进行激酶抑制测定,即分别已知直接参与 Tau 过度磷酸化的 GSK3β、DYRK1A 和 CLK1 受体。有趣的是,生物学评价结果表明化合物IV和V,在芳核中具有溴/氯官能团的优点是对目标 GSK3β 的选择性高于其他。为了加强我们的分析,我们采用了分子建模研究,其中化合物IV/V与参考配体 5-aryl-4-carboxamide-1,3-oxazole 重新停靠在相同的网格 4AFJ 中。令人惊讶的是,我们的研究表明,对于化合物IV/V,设计分子之间存在主要的氢键(IV/V) 和受体 GSK3β 中的 Val 135 残基,与参考配体一致。我们将这种相互作用归因于激发化合物的效力。事实上,衍生物的芳香核与受体 GSK3β 中的 Arg 141/Thr 138 之间的其他非共价相互作用可能是提高靶标选择性的原因。总体而言,我们认为目前的工作描绘了通过系统调整芳环中适当位置的电子密度来微调抑制剂功效的合乎逻辑的证明,无论是主要的喹喔啉还是其他芳环。因此,该途径为开发针对阿尔茨海默氏症等多种神经退行性疾病的有效疗法提供了便利的策略。
更新日期:2021-09-05
中文翻译:
揭示 6,7-二甲基喹喔啉类似物对激酶抑制的选择性:对阿尔茨海默病治疗学发展的见解
通过药物干预解开最具选择性的激酶抑制剂仍然是迄今为止具有挑战性的事务之一。根据这种漂移,本文描述了一组新的杂环类似物的设计和合成,该类似物由附加到连接器的 6,7-二甲基喹喔啉组成,采用席夫碱策略(化合物I – IX)。通过各种光谱技术对化合物进行表征,并对 CMGC 家族的少数主要成员进行激酶抑制测定,即分别已知直接参与 Tau 过度磷酸化的 GSK3β、DYRK1A 和 CLK1 受体。有趣的是,生物学评价结果表明化合物IV和V,在芳核中具有溴/氯官能团的优点是对目标 GSK3β 的选择性高于其他。为了加强我们的分析,我们采用了分子建模研究,其中化合物IV/V与参考配体 5-aryl-4-carboxamide-1,3-oxazole 重新停靠在相同的网格 4AFJ 中。令人惊讶的是,我们的研究表明,对于化合物IV/V,设计分子之间存在主要的氢键(IV/V) 和受体 GSK3β 中的 Val 135 残基,与参考配体一致。我们将这种相互作用归因于激发化合物的效力。事实上,衍生物的芳香核与受体 GSK3β 中的 Arg 141/Thr 138 之间的其他非共价相互作用可能是提高靶标选择性的原因。总体而言,我们认为目前的工作描绘了通过系统调整芳环中适当位置的电子密度来微调抑制剂功效的合乎逻辑的证明,无论是主要的喹喔啉还是其他芳环。因此,该途径为开发针对阿尔茨海默氏症等多种神经退行性疾病的有效疗法提供了便利的策略。


























 京公网安备 11010802027423号
京公网安备 11010802027423号