Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
GWAS for genetics of complex quantitative traits: Genome to pangenome and SNPs to SVs and k-mers
BioEssays ( IF 3.2 ) Pub Date : 2021-09-06 , DOI: 10.1002/bies.202100109 Pushpendra K Gupta 1
BioEssays ( IF 3.2 ) Pub Date : 2021-09-06 , DOI: 10.1002/bies.202100109 Pushpendra K Gupta 1
Affiliation
|
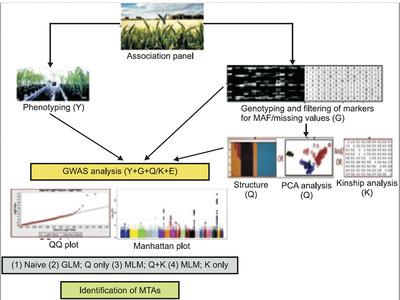
The development of improved methods for genome-wide association studies (GWAS) for genetics of quantitative traits has been an active area of research during the last 25 years. This activity initially started with the use of mixed linear model (MLM), which was variously modified. During the last decade, however, with the availability of high throughput next generation sequencing (NGS) technology, development and use of pangenomes and novel markers including structural variations (SVs) and k-mers for GWAS has taken over as a new thrust area of research. Pangenomes and SVs are now available in humans, livestock, and a number of plant species, so that these resources along with k-mers are being used in GWAS for exploring additional genetic variation that was hitherto not available for analysis. These developments have resulted in significant improvement in GWAS methodology for detection of marker-trait associations (MTAs) that are relevant to human healthcare and crop improvement.
中文翻译:

GWAS 用于复杂数量性状的遗传学:基因组到泛基因组和 SNP 到 SV 和 k-mers
在过去的 25 年里,数量性状遗传学的全基因组关联研究 (GWAS) 的改进方法的开发一直是一个活跃的研究领域。这项活动最初是从使用混合线性模型 (MLM) 开始的,该模型经过各种修改。然而,在过去十年中,随着高通量下一代测序 (NGS) 技术的出现,泛基因组和新标记的开发和使用,包括用于 GWAS 的结构变异 (SV) 和k聚体,已成为研究。泛基因组和 SV 现在可用于人类、牲畜和许多植物物种,因此这些资源与k-mers 正在 GWAS 中用于探索迄今为止无法用于分析的其他遗传变异。这些发展导致 GWAS 方法的显着改进,用于检测与人类医疗保健和作物改良相关的标记性状关联 (MTA)。
更新日期:2021-10-25
中文翻译:
GWAS 用于复杂数量性状的遗传学:基因组到泛基因组和 SNP 到 SV 和 k-mers
在过去的 25 年里,数量性状遗传学的全基因组关联研究 (GWAS) 的改进方法的开发一直是一个活跃的研究领域。这项活动最初是从使用混合线性模型 (MLM) 开始的,该模型经过各种修改。然而,在过去十年中,随着高通量下一代测序 (NGS) 技术的出现,泛基因组和新标记的开发和使用,包括用于 GWAS 的结构变异 (SV) 和k聚体,已成为研究。泛基因组和 SV 现在可用于人类、牲畜和许多植物物种,因此这些资源与k-mers 正在 GWAS 中用于探索迄今为止无法用于分析的其他遗传变异。这些发展导致 GWAS 方法的显着改进,用于检测与人类医疗保健和作物改良相关的标记性状关联 (MTA)。










































 京公网安备 11010802027423号
京公网安备 11010802027423号