当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Time-Resolved Excited-State Analysis of Molecular Electron Dynamics by TDDFT and Bethe–Salpeter Equation Formalisms
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2021-09-06 , DOI: 10.1021/acs.jctc.1c00211 P Grobas Illobre 1 , M Marsili 2 , S Corni 2, 3 , M Stener 1 , D Toffoli 1 , E Coccia 1
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2021-09-06 , DOI: 10.1021/acs.jctc.1c00211 P Grobas Illobre 1 , M Marsili 2 , S Corni 2, 3 , M Stener 1 , D Toffoli 1 , E Coccia 1
Affiliation
|
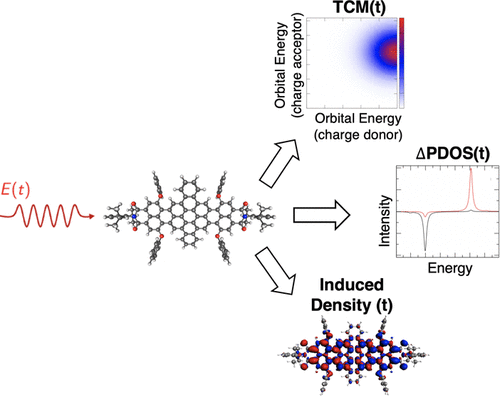
In this work, a theoretical and computational set of tools to study and analyze time-resolved electron dynamics in molecules, under the influence of one or more external pulses, is presented. By coupling electronic-structure methods with the resolution of the time-dependent Schrödinger equation, we developed and implemented the time-resolved induced density of the electronic wavepacket, the time-resolved formulation of the differential projection density of states (ΔPDOS), and of transition contribution map (TCM) to look at the single-electron orbital occupation and localization change in time. Moreover, to further quantify the possible charge transfer, we also defined the energy-integrated ΔPDOS and the fragment-projected TCM. We have used time-dependent density-functional theory (TDDFT), as implemented in ADF software, and the Bethe–Salpeter equation, as provided by MolGW package, for the description of the electronic excited states. This suite of postprocessing tools also provides the time evolution of the electronic states of the system of interest. To illustrate the usefulness of these postprocessing tools, excited-state populations have been computed for HBDI (the chromophore of GFP) and DNQDI molecules interacting with a sequence of two pulses. Time-resolved descriptors have been applied to study the time-resolved electron dynamics of HBDI, DNQDI, LiCN (being a model system for dipole switching upon highest occupied molecular orbital–lowest unoccupied molecular orbital (HOMO–LUMO) electronic excitation), and Ag22. The computational analysis tools presented in this article can be employed to help the interpretation of fast and ultrafast spectroscopies on molecular, supramolecular, and composite systems.
中文翻译:

通过 TDDFT 和 Bethe-Salpeter 方程形式对分子电子动力学进行时间分辨激发态分析
在这项工作中,提出了一组理论和计算工具,用于在一个或多个外部脉冲的影响下研究和分析分子中的时间分辨电子动力学。通过将电子结构方法与时变薛定谔方程的分辨率相结合,我们开发并实现了电子波包的时间分辨感应密度、状态微分投影密度 (ΔPDOS) 的时间分辨公式,以及跃迁贡献图 (TCM) 以查看单电子轨道占据和定位随时间的变化。此外,为了进一步量化可能的电荷转移,我们还定义了能量积分 ΔPDOS 和片段投影 TCM。我们使用了 ADF 软件中实现的时间相关密度泛函理论 (TDDFT),和 Bethe-Salpeter 方程,由 MolGW 包提供,用于描述电子激发态。这套后处理工具还提供了感兴趣系统的电子状态的时间演变。为了说明这些后处理工具的有用性,我们计算了 HBDI(GFP 的生色团)和 DNQDI 分子与两个脉冲序列相互作用的激发态种群。时间分辨描述符已被用于研究 HBDI、DNQDI、LiCN(作为最高占据分子轨道 - 最低未占据分子轨道(HOMO-LUMO)电子激发偶极子切换的模型系统)和 Ag 的时间分辨电子动力学 这套后处理工具还提供了感兴趣系统的电子状态的时间演变。为了说明这些后处理工具的有用性,我们计算了 HBDI(GFP 的生色团)和 DNQDI 分子与两个脉冲序列相互作用的激发态种群。时间分辨描述符已被用于研究 HBDI、DNQDI、LiCN(作为最高占据分子轨道 - 最低未占据分子轨道(HOMO-LUMO)电子激发偶极子切换的模型系统)和 Ag 的时间分辨电子动力学 这套后处理工具还提供了感兴趣系统的电子状态的时间演变。为了说明这些后处理工具的有用性,我们计算了 HBDI(GFP 的生色团)和 DNQDI 分子与两个脉冲序列相互作用的激发态种群。时间分辨描述符已被用于研究 HBDI、DNQDI、LiCN(作为最高占据分子轨道 - 最低未占据分子轨道(HOMO-LUMO)电子激发偶极子切换的模型系统)和 Ag 的时间分辨电子动力学22 . 本文中介绍的计算分析工具可用于帮助解释分子、超分子和复合系统的快速和超快光谱。
更新日期:2021-10-12
中文翻译:
通过 TDDFT 和 Bethe-Salpeter 方程形式对分子电子动力学进行时间分辨激发态分析
在这项工作中,提出了一组理论和计算工具,用于在一个或多个外部脉冲的影响下研究和分析分子中的时间分辨电子动力学。通过将电子结构方法与时变薛定谔方程的分辨率相结合,我们开发并实现了电子波包的时间分辨感应密度、状态微分投影密度 (ΔPDOS) 的时间分辨公式,以及跃迁贡献图 (TCM) 以查看单电子轨道占据和定位随时间的变化。此外,为了进一步量化可能的电荷转移,我们还定义了能量积分 ΔPDOS 和片段投影 TCM。我们使用了 ADF 软件中实现的时间相关密度泛函理论 (TDDFT),和 Bethe-Salpeter 方程,由 MolGW 包提供,用于描述电子激发态。这套后处理工具还提供了感兴趣系统的电子状态的时间演变。为了说明这些后处理工具的有用性,我们计算了 HBDI(GFP 的生色团)和 DNQDI 分子与两个脉冲序列相互作用的激发态种群。时间分辨描述符已被用于研究 HBDI、DNQDI、LiCN(作为最高占据分子轨道 - 最低未占据分子轨道(HOMO-LUMO)电子激发偶极子切换的模型系统)和 Ag 的时间分辨电子动力学 这套后处理工具还提供了感兴趣系统的电子状态的时间演变。为了说明这些后处理工具的有用性,我们计算了 HBDI(GFP 的生色团)和 DNQDI 分子与两个脉冲序列相互作用的激发态种群。时间分辨描述符已被用于研究 HBDI、DNQDI、LiCN(作为最高占据分子轨道 - 最低未占据分子轨道(HOMO-LUMO)电子激发偶极子切换的模型系统)和 Ag 的时间分辨电子动力学 这套后处理工具还提供了感兴趣系统的电子状态的时间演变。为了说明这些后处理工具的有用性,我们计算了 HBDI(GFP 的生色团)和 DNQDI 分子与两个脉冲序列相互作用的激发态种群。时间分辨描述符已被用于研究 HBDI、DNQDI、LiCN(作为最高占据分子轨道 - 最低未占据分子轨道(HOMO-LUMO)电子激发偶极子切换的模型系统)和 Ag 的时间分辨电子动力学22 . 本文中介绍的计算分析工具可用于帮助解释分子、超分子和复合系统的快速和超快光谱。


























 京公网安备 11010802027423号
京公网安备 11010802027423号