Bioorganic Chemistry ( IF 4.5 ) Pub Date : 2021-09-04 , DOI: 10.1016/j.bioorg.2021.105325 Farag F Sherbiny 1 , Ashraf H Bayoumi 2 , Ahmed M El-Morsy 3 , Mohamed Sobhy 2 , Mohamed Hagras 2
|
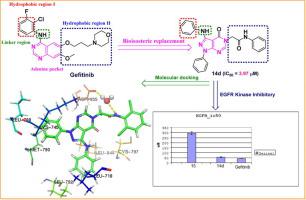
A series of novel hybrid pyrazolo[3,4-d]pyramidine derivatives was designed and chemically synthesized in useful yields. The synthesized compounds were structurally characterized by the usual techniques. All the new synthesized compounds were biologically screened in vitro for their antiproliferative activities against a panel of four cancer cell lines, namely HepG-2, MCF-7, HCT-116, and Hela. The results of cytotoxic evaluation indicated that compound 14d was appeared to be the most prominent broad-spectrum cytotoxic activity and significantly more potent than sorafenib with IC50 values of 4.28, 5.18, 3.97, and 9.85 µM against four cell lines (HePG2, Hela, HCT-116 and MCF-7). In addition, compound 15 was displayed promising antiproliferative effect against all tested cell lines with IC50 value less than 11 µM compared with sorafenib as a control drug. Besides, structurally pharmacophoric features indicated that pyrazolo[3,4-d]pyrimidine scaffold having an amide linker and substituted with phenyl moiety at the 5-position was more potent than those possessing azomethine methyl, azomethine proton and carbomethene linkers, which lead to significant decrease in antiproliferative activity.
The most potent compounds were further selected and evaluated for their activities against epidermal growth factor receptor (EGFR) kinase inhibitors according to homogenous time resolved fluorescence (HTRF) assay. The most potent compound 14d exhibited the most promising inhibitory activity against EGFRWT with IC50 value of 56.02 ± 1.38 µM compared with gefitinib as control drug with IC50 value of 41.79 ± 1.07 µM. Moreover, the inhibition of cell cycle progression and induction of apoptosis in the A549 cell line at G2/M and pre-G1 phases of cell cycle might contribute to cancer treatment that evaluated by Annexin V-FITC/PI double staining detection method. Finally, molecular docking studies were conducted to investigate that probable binding conformations of these anticancer agents and ADME properties were calculated to predict pharmacokinetics and toxic properties of the target compounds.
中文翻译:
新型吡唑并[3,4-d]嘧啶衍生物支架作为有效EGFR抑制剂和细胞凋亡诱导剂的设计、合成、生物学评价和分子对接研究
一系列新型杂化吡唑并 [ 3,4-d ] 嘧啶衍生物被设计并以有用的产率化学合成。合成的化合物通过常规技术进行结构表征。所有新合成的化合物都进行了体外生物筛选,以确定它们对一组四种癌细胞系的抗增殖活性,即 HepG-2、MCF-7、HCT-116 和 Hela。细胞毒性的评价的结果表明,化合物14D是似乎是最突出的广谱的细胞毒活性和显著比用IC索拉非尼更有效的50的4.28的值,5.18,3.97,和9.85μM对四个细胞系(HepG2,Hela细胞, HCT-116 和 MCF-7)。此外,化合物15与作为对照药物的索拉非尼相比,对所有测试细胞系显示出有希望的抗增殖作用,IC 50值小于 11 µM。此外,结构药效特征表明,具有酰胺连接基并在 5-位被苯基部分取代的吡唑并[ 3,4-d ]嘧啶支架比具有甲基偶氮甲碱、偶氮甲碱质子和碳甲烯连接基的那些更有效,从而导致显着的抗增殖活性降低。
根据均相时间分辨荧光 (HTRF) 分析,进一步选择和评估最有效的化合物对表皮生长因子受体 (EGFR) 激酶抑制剂的活性。与作为对照药物的吉非替尼相比,最有效的化合物14d对 EGFR WT表现出最有希望的抑制活性,IC 50值为 56.02 ± 1.38 µM,IC 50值 41.79 ± 1.07 µM。此外,在细胞周期的 G2/M 期和前 G1 期,A549 细胞系中细胞周期进程的抑制和细胞凋亡的诱导可能有助于通过 Annexin V-FITC/PI 双染色检测方法评估的癌症治疗。最后,进行了分子对接研究以研究这些抗癌剂的可能结合构象和 ADME 特性,以预测目标化合物的药代动力学和毒性特性。










































 京公网安备 11010802027423号
京公网安备 11010802027423号