Nano Research ( IF 9.5 ) Pub Date : 2021-09-04 , DOI: 10.1007/s12274-021-3789-x Weiwei Shao 1 , Xiaodong Li 1 , Juncheng Zhu 1 , Xiaolong Zu 1 , Liang Liang 1 , Yongfu Sun 1 , Yi Xie 1 , Jun Hu 2 , Yang Pan 2 , Junfa Zhu 2 , Wensheng Yan 2
|
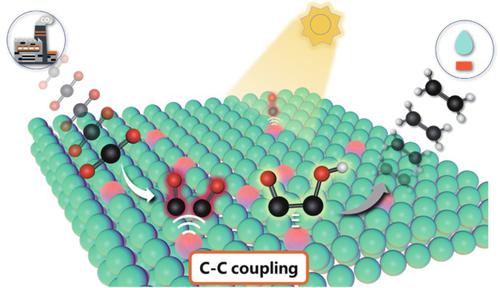
The major obstacle for selective CO2 photoreduction to C2 hydrocarbons lies in the difficulty of C-C coupling, which is usually restrained by the repulsive dipole-dipole interaction between adjacent carbonaceous intermediates. Herein, we first construct semiconducting atomic layers featuring abundant Metaln+-Metalδ+ pair sites (0 < δ < n), aiming to tailor asymmetric charge distribution on the carbonaceous intermediates and hence trigger their C-C coupling for selectively yielding C2 hydrocarbons. As an example, we first fabricate Co-doped NiS2 atomic layers possessing abundant Ni2+-Niδ+ (0 < δ < 2) pairs, where Co doping strategy can ensure higher amount of Ni2+-Niδ+ pair sites. In-situ Fourier-transform infrared spectroscopy, quasi in-situ Raman spectroscopy and density-functional-theory calculations disclose the Ni2+-Niδ+ pair sites endow the adjacent CO intermediates with distinct charge densities, thus decreasing their dipole-dipole repulsion and hence lowering the rate-limiting C-C coupling reaction barrier. As a result, in simulated flue gas (10% CO2 balance 90% N2), the ethylene selectivity for Co-doped NiS2 atomic layers reaches up to 74.3% with an activity of 70 µg·g−1·h−1, outperforming previously reported photocatalysts under similar operating conditions.
中文翻译:
Metaln+-Metalδ+ 对位点引导 CC 耦合以选择性 CO2 光还原为 C2 烃
选择性CO 2 光还原成C 2烃的主要障碍在于CC 耦合的困难,这通常受到相邻碳质中间体之间排斥偶极-偶极相互作用的限制。在此,我们首先构建半导体原子层设有丰富的金属Ñ + -金属δ +对网站(0 < δ < Ñ),旨在裁缝非对称电荷分布在碳质中间体,并因此触发用于选择性地产生Ç其CC联接2烃。例如,我们首先制造了具有丰富 Ni 2+ 的Co 掺杂 NiS 2原子层-Ni δ + (0 < δ < 2) 对,其中 Co 掺杂策略可以确保更高数量的 Ni 2+ -Ni δ +对位点。原位傅立叶变换红外光谱、准原位拉曼光谱和密度泛函理论计算揭示了 Ni 2+ -Ni δ +对位点赋予相邻的 CO 中间体不同的电荷密度,从而降低它们的偶极-偶极排斥从而降低限速 CC 偶联反应势垒。因此,在模拟烟气中(10% CO 2平衡 90% N 2),Co掺杂的 NiS 的乙烯选择性2 个原子层达到 74.3%,活性为 70 µg·g -1 ·h -1,在类似的操作条件下优于先前报道的光催化剂。










































 京公网安备 11010802027423号
京公网安备 11010802027423号