当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
CO2 Adsorptions on d-Block-Metal-Doped Nickel Nanoparticles: Unexpected Adsorption Configurations Predicted by Machine Intelligence
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2021-08-31 , DOI: 10.1021/acs.jpcc.1c07133 Shiru Lin 1 , Chong Teng 1 , Junwei Lucas Bao 1
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2021-08-31 , DOI: 10.1021/acs.jpcc.1c07133 Shiru Lin 1 , Chong Teng 1 , Junwei Lucas Bao 1
Affiliation
|
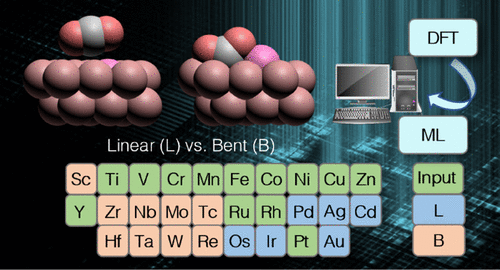
Catalytic CO2 conversion to valuable chemicals on metallic nanoparticles is widely studied in industry. A key step in the thermal activation of adsorbed CO2 is C–O bond scission, which depends sensitively on surface-bound CO2 geometry. However, most of the prior studies used either linear or bent CO2 as the precursor state without considering which geometry is the energetically most stable on a specific metallic surface. The dopant effects on CO2 adsorption geometries were omitted. Herein, we constructed a training set that contains the most stable CO2 adsorption geometries on 12 doped Ni nanoparticles using the Ni (111) facet and trained a classification model for efficient and reliable identification of CO2 adsorption geometry, which predicted unexpected adsorption configurations that were not identified in previous catalytic mechanistic studies. We explored the physical origins of the preferred adsorption geometry and found that the amount of charge transfer between CO2 and the metallic surface plays a critical role.
中文翻译:

d 块金属掺杂镍纳米颗粒上的 CO2 吸附:机器智能预测的意外吸附配置
在金属纳米颗粒上催化 CO 2转化为有价值的化学品在工业中得到了广泛研究。吸附的 CO 2热活化的关键步骤是 C-O 键断裂,它敏感地取决于表面结合的 CO 2几何形状。然而,大多数先前的研究使用线性或弯曲的 CO 2作为前体状态,而没有考虑哪种几何形状在特定金属表面上能量最稳定。省略了掺杂剂对 CO 2吸附几何形状的影响。在这里,我们构建了一个训练集,其中包含最稳定的 CO 2使用 Ni (111) 面对 12 个掺杂的 Ni 纳米粒子进行吸附几何形状,并训练分类模型以有效可靠地识别 CO 2吸附几何形状,该模型预测了在以前的催化机理研究中未发现的意外吸附配置。我们探索了首选吸附几何结构的物理起源,发现 CO 2和金属表面之间的电荷转移量起着关键作用。
更新日期:2021-09-16
中文翻译:
d 块金属掺杂镍纳米颗粒上的 CO2 吸附:机器智能预测的意外吸附配置
在金属纳米颗粒上催化 CO 2转化为有价值的化学品在工业中得到了广泛研究。吸附的 CO 2热活化的关键步骤是 C-O 键断裂,它敏感地取决于表面结合的 CO 2几何形状。然而,大多数先前的研究使用线性或弯曲的 CO 2作为前体状态,而没有考虑哪种几何形状在特定金属表面上能量最稳定。省略了掺杂剂对 CO 2吸附几何形状的影响。在这里,我们构建了一个训练集,其中包含最稳定的 CO 2使用 Ni (111) 面对 12 个掺杂的 Ni 纳米粒子进行吸附几何形状,并训练分类模型以有效可靠地识别 CO 2吸附几何形状,该模型预测了在以前的催化机理研究中未发现的意外吸附配置。我们探索了首选吸附几何结构的物理起源,发现 CO 2和金属表面之间的电荷转移量起着关键作用。










































 京公网安备 11010802027423号
京公网安备 11010802027423号