Journal of Molecular Graphics and Modelling ( IF 2.7 ) Pub Date : 2021-08-28 , DOI: 10.1016/j.jmgm.2021.108012 Roghaye Taherian 1 , Behzad Chahkandi 1 , Ehsan Zahedi 1
|
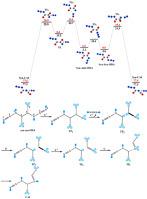
The complete theoretical study of thermal Curtius rearrangement of syn-syn and syn-anti conformers of oxalyl diazide, in the gas phase and in solution has been established for the first time. The inexplicit solvent effect was taken into account via the self-consistent reaction field (SCRF) method. The gas and solution phases of all optimized geometries of the mentioned conformers associated with the Curtius rearrangement along the concerted and stepwise pathways were reported using the polarized continuum model and non-electrostatic terms from the SMD universal solvation model. The Curtius rearrangement of syn-syn and syn-anti conformers was taken place via concerted and stepwise pathways, respectively. The syn-syn conformer of oxalyl diazide is more stable than the syn-anti conformer in the gas phase and solution, and rearranged to syn-carbonyl azide isocyanate via an exergonic concerted mechanism with a single transition state. Nevertheless, the rearrangement of syn-anti conformer occurred through the two transition states and an intermediate, which the first and second steps are endergonic and exergonic, respectively. Theoretical results point out that the concerted pathway is predominant with 102-106 and 104-105 times faster than the stepwise mechanism in gas phase and solution, respectively. Topological analysis of the electron localization function at the B3LYP/6–311++G (2d,d,p) level of theory indicate that the catastrophe sequence 1-6-C†TSC†F C†C-0 begins with the N4–N5 bond breaking, elimination of nitrogen molecule and increasing of non-bonding monosynaptic attractor on N4 atom, and then changing of topological signature of C2–N4 bond, breaking of C1–C2 bond, and formation of pseudo-radical centers on C1 and C2 atoms. Subsequently, annihilation of pseudo-radical centers on the C1 atom, change of topological signature of C2–N4 and formation of C1–N4 bond were executed. The obtained results of ELF calculations show that the reaction takes place via a concerted mechanism but highly asynchronous process.
中文翻译:
草酰二叠氮syn-syn和syn-anti构象异构体的Curtius重排综合理论分析
的热Curtius重排的完整的理论研究顺式顺式和SYN-抗草重氮化物构象,在气相和在溶液中已经建立的第一次。通过自洽反应场 (SCRF) 方法考虑了不明确的溶剂效应。使用极化连续介质模型和来自 SMD 通用溶剂化模型的非静电项报告了与沿协调和逐步路径的 Curtius 重排相关的上述构象异构体的所有优化几何结构的气相和溶液相。Syn -syn和syn-anti构象异构体的 Curtius 重排分别通过协调和逐步途径发生。该SYN-SYN草酰二叠氮的构象异构体在气相和溶液中比顺反构象异构体更稳定,并通过具有单一过渡态的放能协调机制重排为顺-羰基叠氮化物异氰酸酯。然而,syn-anti 构象异构体的重排通过两个过渡态和一个中间体发生,其中第一步和第二步分别是 endergonic 和 exergonic。理论结果表明,在气相和溶液中,协同途径占优势,分别比逐步机制快10 2 -10 6和10 4 -10 5倍。B3LYP/6–311++G 电子定位函数的拓扑分析(2d,d,p ) 理论水平表明灾难序列 1-6-C †TS C † FC † C-0 从 N 4 –N 5键断裂、氮分子消除和非键单突触增加开始N 4原子上的吸引子,然后改变C 2 -N 4键的拓扑特征,破坏C 1 -C 2键,并在C 1和C 2原子上形成伪自由基中心。随后,C 1原子上的伪自由基中心湮灭,C 2 –N的拓扑特征发生变化4并形成C 1 -N 4键。ELF 计算的结果表明,反应是通过协同机制但高度异步的过程发生的。










































 京公网安备 11010802027423号
京公网安备 11010802027423号