Carbon ( IF 10.5 ) Pub Date : 2021-08-29 , DOI: 10.1016/j.carbon.2021.08.070 Rhea Montgomery-Walsh 1, 2 , Surabhi Nimbalkar 1, 2 , James Bunnell 1, 2 , Sandra Lara Galindo 1, 2 , Sam Kassegne 1, 2
|
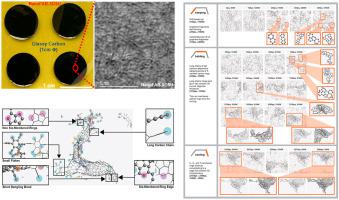
With the continued and sustained growth of the field of carbon MEMS (C-MEMS) in several application areas, there is a corresponding increased and renewed interest in understanding the exact molecular structure of the ensuing glassy carbon (GC) material and, therefore, potential to engineer some of its key characteristics. Associated with that interest, the effects of key process parameters like temperature ramping, holding and cooling times on carbonization, the final nanostructures and – importantly – on the nature and distribution of functional groups remain to be of significant research importance to the GC community. While recent in-situ imaging approaches during the synthesis process have opened up opportunities for better understanding of these nanostructures, the 2D nature of the approaches, however, had introduced significant barriers. In this paper, therefore, we present a simulation-based approach using reactive molecular dynamics (MD) where we model the synthesis process and investigate in detail (i) the intermediate and final stages of carbonization with particular focus on GC nanostructures and (ii) formation of functional groups and the processes and parameters that drive their evolution. Our simulation captured the pyrolysis-driven synthesis process where the polymer structure degraded followed by increased carbonization and formation of flakes of 3-, 4-, membered carbon rings that further grew into graphene-type 6-member carbon rings as well as 5- and 7-member rings. These eventually evolved into a complex multi-layer 3D nanostructure consisting of connected but distinct zones of (a) dominant cage-like tubular components and (b) long chains of flat graphene-like parts rich with functional groups. Taken together, the MD simulations demonstrate that (i) holding time at the maximum temperature has the most significant effect in the formation of complex cage-like 3D nanostructures with stacked layers, (ii) the closed cage-like zones have crystallite diameter (La) in the range of 3.2 Å - 9.5 Å with clear and well-defined curved and stacked graphene-like sheets of stacking thickness (Lc) = 3.0 Å - 3.3 Å, (iii) carbonyl, ether, epoxide, hydroxyl, and alkyne groups form predominately on the edges of flat components of GC where abundant non-6 membered rings, dangling bonds, long sp carbon chains, and intermediate structures exist, and to a lesser extent on edges of vacancy defects inside cage-like zones.
中文翻译:
玻璃碳热解作用下纳米结构和官能团演化的分子动力学模拟
随着碳微机电系统 (C-MEMS) 领域在多个应用领域的持续和持续增长,人们对了解后续玻璃碳 (GC) 材料的确切分子结构的兴趣也相应增加并重新产生兴趣,因此,潜在的设计它的一些关键特性。与这种兴趣相关的是,诸如温度上升、保温和冷却时间等关键工艺参数对碳化、最终纳米结构以及——重要的是——对官能团的性质和分布的影响对于 GC 社区仍然具有重要的研究意义。虽然最近在现场合成过程中的成像方法为更好地理解这些纳米结构开辟了机会,然而,这些方法的二维性质带来了重大障碍。因此,在本文中,我们提出了一种使用反应性分子动力学 (MD) 的基于模拟的方法,其中我们对合成过程进行建模并详细研究 (i) 碳化的中间和最后阶段,特别关注 GC 纳米结构和 (ii)功能组的形成以及驱动其进化的过程和参数。我们的模拟捕获了热解驱动的合成过程,其中聚合物结构降解,随后碳化增加并形成 3-、4- 元碳环薄片,进一步生长成石墨烯型 6-元碳环以及 5-和7人戒指。这些最终演变成复杂的多层 3D 纳米结构,由连接但不同的区域组成(a)主要的笼状管状成分和(b)富含官能团的扁平石墨烯状部分的长链。综上所述,MD 模拟表明(i)在最高温度下的保持时间对形成具有堆叠层的复杂笼状 3D 纳米结构的影响最显着,(ii)封闭的笼状区域具有微晶直径(La ) 范围内3.2 Å - 9.5 Å具有清晰且定义明确的弯曲和堆叠石墨烯状片,堆叠厚度 (Lc) = 3.0 Å - 3.3 Å,(iii) 羰基、醚、环氧化物、羟基和炔基主要在边缘形成GC 的扁平组分,其中存在丰富的非 6 元环、悬空键、长sp碳链和中间结构,并且在较小程度上存在于笼状区域内的空位缺陷边缘。










































 京公网安备 11010802027423号
京公网安备 11010802027423号