当前位置:
X-MOL 学术
›
J. Phys. Org. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Theoretical investigations on mechanisms and kinetics of methylketene with H reaction in the atmosphere
Journal of Physical Organic Chemistry ( IF 1.9 ) Pub Date : 2021-08-27 , DOI: 10.1002/poc.4274 Huaming Du 1 , Yuxi Sun 1 , Huirong Li 1 , Zhiguo Wang 1 , Yunju Zhang 1, 2 , Yongguo Liu 2 , Meilian Zhao 3
Journal of Physical Organic Chemistry ( IF 1.9 ) Pub Date : 2021-08-27 , DOI: 10.1002/poc.4274 Huaming Du 1 , Yuxi Sun 1 , Huirong Li 1 , Zhiguo Wang 1 , Yunju Zhang 1, 2 , Yongguo Liu 2 , Meilian Zhao 3
Affiliation
|
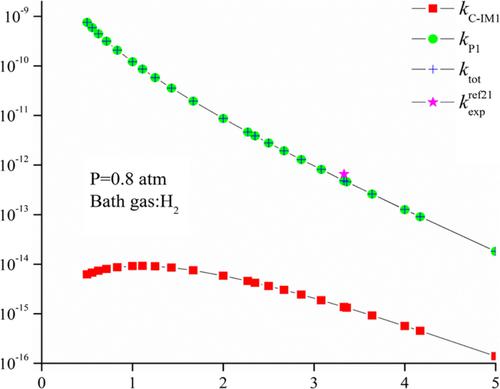
Detailed oxidation course of H-initiated degradation of CH3CHCO has been researched taking advantage of quantum chemical methods. Geometrical optimizations of reactants, intermediates, transition states, and products were operated at the B3LYP/6-31++G(d,p) level. Single-point energy computations were implemented at the CCSD(T)/cc-pVTZ//B3LYP/6-311++G(d,p) level. The results state clearly that the H association was more energetically beneficial than the abstraction of H, and the dominant pathway is generation of P1 (CH3CH2 + CO). Rate constants of H association reactions are computed by making use of Rice–Ramsperger–Kassel–Marcus (RRKM) program at 200–3000 K. Specifically, the total rate constant of H association reactions is 6.47 × 10−13 cm3 molecule−1 s−1 at 298 K, which is consistent with the experimental results (6.58 × 10−13 cm3 molecule−1 s−1). This research offers a thorough comprehending of the reaction mechanism involved in H-initiated atmospheric degradation of CH3CHCO and might act as a relevant supplemental reference criteria for experimental research.
中文翻译:

甲基乙烯酮在大气中与H反应的机理和动力学的理论研究
利用量子化学方法研究了H引发降解CH 3 CHCO的详细氧化过程。反应物、中间体、过渡态和产物的几何优化在 B3LYP/6-31++G(d,p) 水平上进行。单点能量计算在 CCSD(T)/cc-pVTZ//B3LYP/6-311++G(d,p) 水平上实现。结果清楚地表明,H 关联比 H 的提取在能量上更有益,主要途径是 P1 (CH 3 CH 2 + CO) 的生成。H 缔合反应的速率常数通过利用 Rice-Ramsperger-Kassel-Marcus (RRKM) 程序在 200-3000 K 下计算。具体而言,H 缔合反应的总速率常数为 6.47 × 10 -13 cm 3 分子-1 s -1在 298 K,这与实验结果(6.58 × 10 -13 cm 3 分子-1 s -1)一致。本研究深入理解了氢气引发的 CH 3 CHCO大气降解反应机理,可作为实验研究的相关补充参考标准。
更新日期:2021-08-27
中文翻译:
甲基乙烯酮在大气中与H反应的机理和动力学的理论研究
利用量子化学方法研究了H引发降解CH 3 CHCO的详细氧化过程。反应物、中间体、过渡态和产物的几何优化在 B3LYP/6-31++G(d,p) 水平上进行。单点能量计算在 CCSD(T)/cc-pVTZ//B3LYP/6-311++G(d,p) 水平上实现。结果清楚地表明,H 关联比 H 的提取在能量上更有益,主要途径是 P1 (CH 3 CH 2 + CO) 的生成。H 缔合反应的速率常数通过利用 Rice-Ramsperger-Kassel-Marcus (RRKM) 程序在 200-3000 K 下计算。具体而言,H 缔合反应的总速率常数为 6.47 × 10 -13 cm 3 分子-1 s -1在 298 K,这与实验结果(6.58 × 10 -13 cm 3 分子-1 s -1)一致。本研究深入理解了氢气引发的 CH 3 CHCO大气降解反应机理,可作为实验研究的相关补充参考标准。










































 京公网安备 11010802027423号
京公网安备 11010802027423号