当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Interplay of the Ring and Steric Strains in the Highly Substituted Cyclopropanes
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2021-08-25 , DOI: 10.1021/acs.jpca.1c04777 Sergey L Khursan 1 , Ekaterina S Akhmetshina 1
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2021-08-25 , DOI: 10.1021/acs.jpca.1c04777 Sergey L Khursan 1 , Ekaterina S Akhmetshina 1
Affiliation
|
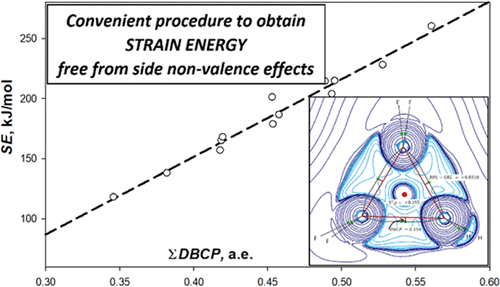
Using the concept of a complete set of homodesmotic reactions for the analysis of molecular energetics of polysubstituted methyl- and fluorocyclopropanes allows assessing the strain energy SE of cyclopropanes, free from interfering effects, in full accordance with the IUPAC definition (“relative to a reference ... hypothetical ‘strainless’ structure”). The correct SE calculation requires quantifying nonvalence interactions in the products of formal homodesmotic reactions (HDRs) using a routine multiregression analysis. The complete HDR set provides the information necessary for the analysis, namely, the heat effects of HDRs calculated by the G4 composite method and the wide set of reference compounds with various combinations of nonvalence effects. We have found that the SE value for methylcyclopropanes lies in the range from 117.0 (1.1-dimethylcyclopropane) to 146.1 kJ/mol (hexamethylcyclopropane). It is the sum of the ring strain energy RSE = 117.9 ± 0.3 kJ mol, which does not depend on the number of methyl substituents, and the Pitzer strain energy of 4.4±0.1 kJ/mol per one contact (the standard deviation is shown as an error of determination). In the series of fluorocyclopropanes, SE varies from 137.9 (monosubstituted cyclopropane) to 260.0 kJ/mol (hexafluorocyclopropane) and well correlates with the ∑DBCP parameter deduced from the QTAIM analysis of the electron density of the compound, representing the total deviation of bond critical points from geometrical C–C bond lines of CC bonds. The ∑DBCP parameter characterizes the curvature of banana-like bonds in cyclopropanes.
中文翻译:

高度取代的环丙烷中环和空间应变的相互作用
使用一套完整的同质反应的概念来分析多取代甲基和氟环丙烷的分子能量学,可以评估环丙烷的应变能 SE,不受干扰影响,完全符合 IUPAC 定义(“相对于参考 . .. 假设的“无应变”结构”)。正确的 SE 计算需要使用常规多元回归分析量化形式同质反应 (HDR) 产物中的非价相互作用。完整的 HDR 集提供了分析所需的信息,即通过 G4 复合方法计算的 HDR 的热效应和具有各种非价效应组合的广泛参考化合物集。我们发现甲基环丙烷的 SE 值在 117.0 (1. 1-二甲基环丙烷)至 146.1 kJ/mol(六甲基环丙烷)。它是环应变能 RSE = 117.9 ± 0.3 kJ mol(不依赖于甲基取代基的数量)与每一次接触的 Pitzer 应变能 4.4±0.1 kJ/mol 之和(标准偏差表示为判断错误)。在氟环丙烷系列中,SE 从 137.9(单取代环丙烷)到 260.0 kJ/mol(六氟环丙烷)变化,并且与从化合物电子密度的 QTAIM 分析推导出的 ∑DBCP 参数很好地相关,代表键临界的总偏差CC 键的几何 C-C 键合线的点。∑DBCP 参数表征了环丙烷中香蕉状键的曲率。9 ± 0.3 kJ mol,不依赖于甲基取代基的数量,每次接触的 Pitzer 应变能为 4.4±0.1 kJ/mol(标准偏差表示为测定误差)。在氟环丙烷系列中,SE 从 137.9(单取代环丙烷)到 260.0 kJ/mol(六氟环丙烷)变化,并且与从化合物电子密度的 QTAIM 分析推导出的 ∑DBCP 参数很好地相关,代表键临界的总偏差CC 键的几何 C-C 键合线的点。∑DBCP 参数表征了环丙烷中香蕉状键的曲率。9 ± 0.3 kJ mol,不依赖于甲基取代基的数量,每次接触的 Pitzer 应变能为 4.4±0.1 kJ/mol(标准偏差表示为测定误差)。在氟环丙烷系列中,SE 从 137.9(单取代环丙烷)到 260.0 kJ/mol(六氟环丙烷)变化,并且与从化合物电子密度的 QTAIM 分析推导出的 ∑DBCP 参数很好地相关,代表键临界的总偏差CC 键的几何 C-C 键合线的点。∑DBCP 参数表征了环丙烷中香蕉状键的曲率。在氟环丙烷系列中,SE 从 137.9(单取代环丙烷)到 260.0 kJ/mol(六氟环丙烷)变化,并且与从化合物电子密度的 QTAIM 分析推导出的 ∑DBCP 参数很好地相关,代表键临界的总偏差CC 键的几何 C-C 键合线的点。∑DBCP 参数表征了环丙烷中香蕉状键的曲率。在氟环丙烷系列中,SE 从 137.9(单取代环丙烷)到 260.0 kJ/mol(六氟环丙烷)变化,并且与从化合物电子密度的 QTAIM 分析推导出的 ∑DBCP 参数很好地相关,代表键临界的总偏差CC 键的几何 C-C 键合线的点。∑DBCP 参数表征了环丙烷中香蕉状键的曲率。
更新日期:2021-09-09
中文翻译:
高度取代的环丙烷中环和空间应变的相互作用
使用一套完整的同质反应的概念来分析多取代甲基和氟环丙烷的分子能量学,可以评估环丙烷的应变能 SE,不受干扰影响,完全符合 IUPAC 定义(“相对于参考 . .. 假设的“无应变”结构”)。正确的 SE 计算需要使用常规多元回归分析量化形式同质反应 (HDR) 产物中的非价相互作用。完整的 HDR 集提供了分析所需的信息,即通过 G4 复合方法计算的 HDR 的热效应和具有各种非价效应组合的广泛参考化合物集。我们发现甲基环丙烷的 SE 值在 117.0 (1. 1-二甲基环丙烷)至 146.1 kJ/mol(六甲基环丙烷)。它是环应变能 RSE = 117.9 ± 0.3 kJ mol(不依赖于甲基取代基的数量)与每一次接触的 Pitzer 应变能 4.4±0.1 kJ/mol 之和(标准偏差表示为判断错误)。在氟环丙烷系列中,SE 从 137.9(单取代环丙烷)到 260.0 kJ/mol(六氟环丙烷)变化,并且与从化合物电子密度的 QTAIM 分析推导出的 ∑DBCP 参数很好地相关,代表键临界的总偏差CC 键的几何 C-C 键合线的点。∑DBCP 参数表征了环丙烷中香蕉状键的曲率。9 ± 0.3 kJ mol,不依赖于甲基取代基的数量,每次接触的 Pitzer 应变能为 4.4±0.1 kJ/mol(标准偏差表示为测定误差)。在氟环丙烷系列中,SE 从 137.9(单取代环丙烷)到 260.0 kJ/mol(六氟环丙烷)变化,并且与从化合物电子密度的 QTAIM 分析推导出的 ∑DBCP 参数很好地相关,代表键临界的总偏差CC 键的几何 C-C 键合线的点。∑DBCP 参数表征了环丙烷中香蕉状键的曲率。9 ± 0.3 kJ mol,不依赖于甲基取代基的数量,每次接触的 Pitzer 应变能为 4.4±0.1 kJ/mol(标准偏差表示为测定误差)。在氟环丙烷系列中,SE 从 137.9(单取代环丙烷)到 260.0 kJ/mol(六氟环丙烷)变化,并且与从化合物电子密度的 QTAIM 分析推导出的 ∑DBCP 参数很好地相关,代表键临界的总偏差CC 键的几何 C-C 键合线的点。∑DBCP 参数表征了环丙烷中香蕉状键的曲率。在氟环丙烷系列中,SE 从 137.9(单取代环丙烷)到 260.0 kJ/mol(六氟环丙烷)变化,并且与从化合物电子密度的 QTAIM 分析推导出的 ∑DBCP 参数很好地相关,代表键临界的总偏差CC 键的几何 C-C 键合线的点。∑DBCP 参数表征了环丙烷中香蕉状键的曲率。在氟环丙烷系列中,SE 从 137.9(单取代环丙烷)到 260.0 kJ/mol(六氟环丙烷)变化,并且与从化合物电子密度的 QTAIM 分析推导出的 ∑DBCP 参数很好地相关,代表键临界的总偏差CC 键的几何 C-C 键合线的点。∑DBCP 参数表征了环丙烷中香蕉状键的曲率。










































 京公网安备 11010802027423号
京公网安备 11010802027423号