Surface Science ( IF 2.1 ) Pub Date : 2021-08-20 , DOI: 10.1016/j.susc.2021.121931 Novianto Nur Hidayat 1 , Mohammad Kemal Agusta 2, 3 , Hermawan Kresno Dipojono 2, 3 , Hiroshi Nakanishi 4 , Hideaki Kasai 1, 4
|
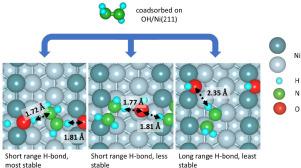
Hydrazine (N2H4) and hydroxyl (OH) coadsorption on the Ni(211) surface is investigated using Density Functional Theory (DFT) based calculations. Effects of the vicinal steps and edges on the stability of various N2H4-OH coadsorption configurations are investigated and compared with the case of individual adsorption of the respective molecules. Stabilizing interaction between N2H4 and OH coadsorbate is found to be mediated by hydrogen-bond. The most stable interaction of N2H4-OH is found perpendicular to the step direction, with N2H4 and OH occupying the terrace and edge sites, respectively. Swapping the position of N2H4 and OH reduce the relative stability. However, placing N2H4 on edge sites stabilizes its cis-conformation with noticeable N - H bond elongation. Finally, N2H4 and OH coadsorption with N2H4 and OH coadsorbed on edge-sites is moderate in terms of overall stability but exhibits the weakest N2H4 coadsorption and longest hydrogen bond interaction distance. Nevertheless, these N2H4-OH coadsorption structures are almost equally accessible thermodynamically due to the relatively small energy differences among them (less than 0.2 eV). In an ideal extended periodic system at specific coverage used in this study, the obtained coadsorption reveals the formation of a chain-like structure between N2H4-OH linked by hydrogen bonds. The results demonstrate the importance of steps and edges in effective formations of hydrogen bond stabilizing N2H4 coadsorption with prospects of promoting proton-transfer-reaction from N2H4 to OH.
中文翻译:
Ni(211) 表面上肼和 OH 的共吸附:DFT 研究
使用基于密度泛函理论 (DFT) 的计算研究了 Ni(211) 表面上的肼 (N 2 H 4 ) 和羟基 (OH) 共吸附。研究了相邻台阶和边缘对各种 N 2 H 4 -OH 共吸附构型稳定性的影响,并与各个分子单独吸附的情况进行了比较。发现N 2 H 4和 OH 共吸附物之间的稳定相互作用是由氢键介导的。发现N 2 H 4 -OH最稳定的相互作用垂直于台阶方向,其中 N 2 H 4和 OH 分别占据平台和边缘站点。交换N 2 H 4和OH 的位置会降低相对稳定性。然而,将 N 2 H 4置于边缘位点可稳定其顺式构象,并具有显着的 N - H 键伸长。最后,N 2 H 4和OH 共吸附,N 2 H 4和OH 共吸附在边缘位点上,总体稳定性适中,但N 2 H 4 共吸附最弱,氢键相互作用距离最长。尽管如此,这些 N 2 H 4由于它们之间的能量差异相对较小(小于 0.2 eV),-OH 共吸附结构在热力学上几乎相同。在本研究中使用的特定覆盖范围的理想扩展周期系统中,获得的共吸附揭示了通过氢键连接的N 2 H 4 -OH之间形成链状结构。结果证明了台阶和边缘在稳定 N 2 H 4 共吸附的氢键的有效形成中的重要性,具有促进从 N 2 H 4到 OH 的质子转移反应的前景。










































 京公网安备 11010802027423号
京公网安备 11010802027423号