Materials Today ( IF 21.1 ) Pub Date : 2021-08-20 , DOI: 10.1016/j.mattod.2021.05.021 Xin-Gang Zhao 1 , Zhi Wang 1, 2 , Oleksandr I. Malyi 1 , Alex Zunger 1
|
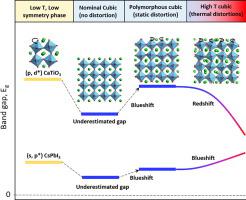
Ternary ABX3 perovskites made of corner-sharing BX6 octahedra have long featured prominently in solid-state chemistry and condensed matter physics. Still, the joint understanding of their two main subgroups—halides and oxides—has not been fully developed. Indeed, unlike the case in simpler compounds having a single, robust repeated motif (“monomorphous”), certain cubic perovskites can manifest a non-thermal (= intrinsic) distribution of local motifs (“polymorphous networks”). Such static deformations can include positional degrees of freedom (e.g., atomic displacements and octahedral tilting) or magnetic moment degrees of freedom in paramagnets. Unlike thermal motion, such static distortions do not time-average to zero, being an expression of the intrinsic symmetry breaking preference of the chemical bonding. The present study compares electronic structure features of oxide and halide perovskites starting from the static polymorphous distribution of motifs described by Density Functional Theory (DFT) minimization of the internal energy, continuing to finite temperature thermal disorder modeled via finite temperature DFT molecular dynamics. We find that (i) different oxide vs. halide ABX3 compounds adopt different energy-lowering symmetry-breaking modes. The calculated pair distribution function (PDF) of SrTiO3 from the first-principles agrees with recently measured PDF. (ii) In both oxides and halides, such static distortions lead to band gap blueshifts with respect to undistorted cubic Pm-3m structure. (iii) For oxide perovskites, high-temperature molecular dynamics simulations initiated from the statically distorted polymorphous structures reveal that the thermally-induced distortions can lead to a band gap redshift. (iv) In contrast, for cubic halide perovskite CsPbI3, both the intrinsic distortions and the thermal distortions contribute in tandem to band gap blueshift, the former, intrinsic effect being dominant. (v) In the oxide SrTiO3 and CaTiO3 (but not in halide) perovskites, octahedral tilting leads to the emergence of a distinct Γ–Γ direct band gap component as a secondary valley minimum to the well-known indirect R–Γ gap. Understanding such intrinsic vs. thermal effects on oxide vs. halide perovskites holds the potential for designing target electronic properties.
中文翻译:
静态局部扭曲与动态运动对立方氧化物和卤化物钙钛矿稳定性和带隙的影响
由共享角的 BX 6八面体制成的三元 ABX 3钙钛矿长期以来一直在固态化学和凝聚态物理中占有突出地位。尽管如此,对它们的两个主要亚群——卤化物和氧化物的共同理解——尚未完全开发。事实上,与具有单一、稳健的重复基序(“单晶”)的更简单化合物的情况不同,某些立方钙钛矿可以表现出局部基序的非热(= 内在)分布(“多晶网络”)。这种静态变形可以包括顺磁体中的位置自由度(例如,原子位移和八面体倾斜)或磁矩自由度。与热运动不同,这种静态扭曲的时间平均不会为零,这是化学键的固有对称性破坏偏好的表达。本研究比较了氧化物和卤化物钙钛矿的电子结构特征,从内部能量的密度泛函理论 (DFT) 最小化描述的基序的静态多态分布开始,继续通过有限温度 DFT 分子动力学建模的有限温度热无序。我们发现 (i) 不同氧化物与卤化物ABX 3化合物采用不同的降低能量的对称破坏模式。根据第一性原理计算的 SrTiO 3 的对分布函数 (PDF)与最近测量的 PDF 一致。(ii) 在氧化物和卤化物中,这种静态畸变导致相对于未畸变的立方Pm -3 m结构的带隙蓝移。(iii) 对于氧化物钙钛矿,从静态扭曲的多晶结构开始的高温分子动力学模拟表明,热诱导的扭曲会导致带隙红移。(iv) 相反,对于立方卤化物钙钛矿 CsPbI如图 3 所示,内在畸变和热畸变共同导致带隙蓝移,前者内在效应占主导地位。(v) 在氧化物 SrTiO 3和 CaTiO 3(但不是在卤化物中)钙钛矿中,八面体倾斜导致出现明显的 Γ-Γ 直接带隙分量作为众所周知的间接 R-Γ 间隙的次要谷最小值. 了解这种对氧化物与卤化物钙钛矿的固有与热效应具有设计目标电子特性的潜力。










































 京公网安备 11010802027423号
京公网安备 11010802027423号