当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Combining machine learning and quantum mechanics yields more chemically aware molecular descriptors for medicinal chemistry applications
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2021-08-19 , DOI: 10.1002/jcc.26737 Sara Tortorella 1 , Emanuele Carosati 2 , Giulia Sorbi 1 , Giovanni Bocci 3 , Simon Cross 4 , Gabriele Cruciani 2 , Loriano Storchi 4, 5
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2021-08-19 , DOI: 10.1002/jcc.26737 Sara Tortorella 1 , Emanuele Carosati 2 , Giulia Sorbi 1 , Giovanni Bocci 3 , Simon Cross 4 , Gabriele Cruciani 2 , Loriano Storchi 4, 5
Affiliation
|
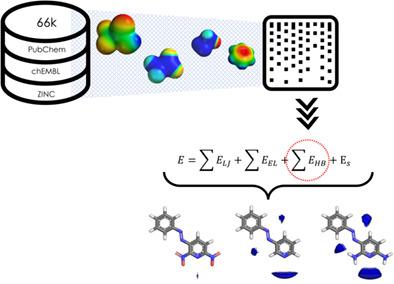
Molecular interaction fields (MIFs), describing molecules in terms of their ability to interact with any chemical entity, are one of the most established and versatile concepts in drug discovery. Improvement of this molecular description is highly desirable for in silico drug discovery and medicinal chemistry applications. In this work, we revised a well-established molecular mechanics' force field and applied a hybrid quantum mechanics and machine learning approach to parametrize the hydrogen-bonding (HB) potentials of small molecules, improving this aspect of the molecular description. Approximately 66,000 molecules were chosen from available drug databases and subjected to density functional theory calculations (DFT). For each atom, the molecular electrostatic potential (EP) was extracted and used to derive new HB energy contributions; this was subsequently combined with a fingerprint-based description of the structural environment via partial least squares modeling, enabling the new potentials to be used for molecules outside of the training set. We demonstrate that parameter prediction for molecules outside of the training set correlates with their DFT-derived EP, and that there is correlation of the new potentials with hydrogen-bond acidity and basicity scales. We show the newly derived MIFs vary in strength for various ring substitution in accordance with chemical intuition. Finally, we report that this derived parameter, when extended to non-HB atoms, can also be used to estimate sites of reaction.
中文翻译:

结合机器学习和量子力学为药物化学应用产生更多化学感知分子描述符
分子相互作用场 (MIF),根据分子与任何化学实体相互作用的能力来描述分子,是药物发现中最成熟和最通用的概念之一。这种分子描述的改进对于计算机药物发现和药物化学应用是非常可取的。在这项工作中,我们修改了成熟的分子力学力场,并应用混合量子力学和机器学习方法来参数化小分子的氢键 (HB) 势,从而改进分子描述的这一方面。从可用的药物数据库中选择了大约 66,000 个分子,并进行了密度泛函理论计算 (DFT)。对于每个原子,提取分子静电势 (EP) 并用于推导新的 HB 能量贡献;随后通过偏最小二乘建模将其与基于指纹的结构环境描述相结合,使新的潜力可用于训练集之外的分子。我们证明了训练集之外的分子的参数预测与其 DFT 衍生的 EP 相关,并且新的势能与氢键酸碱度尺度相关。我们展示了新衍生的 MIF 根据化学直觉在各种环取代的强度上有所不同。最后,我们报告这个派生参数,当扩展到非 HB 原子时,也可用于估计反应位点。使新的潜力可用于训练集之外的分子。我们证明了训练集之外的分子的参数预测与其 DFT 衍生的 EP 相关,并且新的势能与氢键酸碱度尺度相关。我们展示了新衍生的 MIF 根据化学直觉在各种环取代的强度上有所不同。最后,我们报告这个派生参数,当扩展到非 HB 原子时,也可用于估计反应位点。使新的潜力可用于训练集之外的分子。我们证明了训练集之外的分子的参数预测与其 DFT 衍生的 EP 相关,并且新的势能与氢键酸碱度尺度相关。我们展示了新衍生的 MIF 根据化学直觉在各种环取代的强度上有所不同。最后,我们报告这个派生参数,当扩展到非 HB 原子时,也可用于估计反应位点。我们展示了新衍生的 MIF 根据化学直觉在各种环取代的强度上有所不同。最后,我们报告这个派生参数,当扩展到非 HB 原子时,也可用于估计反应位点。我们展示了新衍生的 MIF 根据化学直觉在各种环取代的强度上有所不同。最后,我们报告这个派生参数,当扩展到非 HB 原子时,也可用于估计反应位点。
更新日期:2021-09-24
中文翻译:
结合机器学习和量子力学为药物化学应用产生更多化学感知分子描述符
分子相互作用场 (MIF),根据分子与任何化学实体相互作用的能力来描述分子,是药物发现中最成熟和最通用的概念之一。这种分子描述的改进对于计算机药物发现和药物化学应用是非常可取的。在这项工作中,我们修改了成熟的分子力学力场,并应用混合量子力学和机器学习方法来参数化小分子的氢键 (HB) 势,从而改进分子描述的这一方面。从可用的药物数据库中选择了大约 66,000 个分子,并进行了密度泛函理论计算 (DFT)。对于每个原子,提取分子静电势 (EP) 并用于推导新的 HB 能量贡献;随后通过偏最小二乘建模将其与基于指纹的结构环境描述相结合,使新的潜力可用于训练集之外的分子。我们证明了训练集之外的分子的参数预测与其 DFT 衍生的 EP 相关,并且新的势能与氢键酸碱度尺度相关。我们展示了新衍生的 MIF 根据化学直觉在各种环取代的强度上有所不同。最后,我们报告这个派生参数,当扩展到非 HB 原子时,也可用于估计反应位点。使新的潜力可用于训练集之外的分子。我们证明了训练集之外的分子的参数预测与其 DFT 衍生的 EP 相关,并且新的势能与氢键酸碱度尺度相关。我们展示了新衍生的 MIF 根据化学直觉在各种环取代的强度上有所不同。最后,我们报告这个派生参数,当扩展到非 HB 原子时,也可用于估计反应位点。使新的潜力可用于训练集之外的分子。我们证明了训练集之外的分子的参数预测与其 DFT 衍生的 EP 相关,并且新的势能与氢键酸碱度尺度相关。我们展示了新衍生的 MIF 根据化学直觉在各种环取代的强度上有所不同。最后,我们报告这个派生参数,当扩展到非 HB 原子时,也可用于估计反应位点。我们展示了新衍生的 MIF 根据化学直觉在各种环取代的强度上有所不同。最后,我们报告这个派生参数,当扩展到非 HB 原子时,也可用于估计反应位点。我们展示了新衍生的 MIF 根据化学直觉在各种环取代的强度上有所不同。最后,我们报告这个派生参数,当扩展到非 HB 原子时,也可用于估计反应位点。










































 京公网安备 11010802027423号
京公网安备 11010802027423号