Computational Materials Science ( IF 3.1 ) Pub Date : 2021-08-19 , DOI: 10.1016/j.commatsci.2021.110787 Jing Guo 1 , Bin Xiao 2 , Yihang Li 2 , Dong Zhai 3 , Yuchao Tang 1 , Wan Du 2 , Yi Liu 1, 2
|
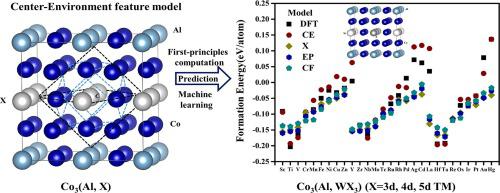
To understand the alloying effects on the stability of Co3Al precipitate phase in Co-based superalloy, the energetic stability and structure of ternary alloy Co3(Al, X) doped with the thirty 3d, 4d, and 5d transition metal (TM) elements were studied in this work using first-principles (FP) computation and machine learning (ML) methods. Our FP computation indicated that Hf, Ta, and Ti doping were thermodynamically most stable. Based on the FP computation data, the ML models with three types of chemical composition (CC) and Center-Environment (CE) features, were developed to predict the formation energies and lattice constants of Co3(Al, X) (X = 3d, 4d, and 5d TM elements). The results show that these ML models all had good prediction accuracy with averaged mean absolute errors (<MAE > ) ~ 0.02 eV/atom for formation energies and ~ 0.01 Å for lattice constants. Then, the effects of important features were discussed on the energy and geometry properties. To study the alloying effects on the stability of Co3(Al, W) precipitate phase, the ML models of Co3(Al, X) were found to be equally accurate to predict the untrained structures of Co3(Al, WX3) where X = 3d, 4d, and 5d TM elements excluding Co and W. We found that the Co3(Al, WX3) structures became most stable when X are IVB and VB group TM elements. This work show that machine learning methods can efficiently extend the capabilities of first-principles predictions on the structure stabilities in multi-component alloy design.
中文翻译:
机器学习辅助过渡金属元素掺杂 Co3(Al, X) 结构稳定性的第一性原理研究
了解合金化对Co 基高温合金中 Co 3 Al 析出相稳定性的影响,以及掺杂 30 种 3d、4d 和 5d 过渡金属 (TM)的三元合金 Co 3 (Al, X)的能量稳定性和结构在这项工作中使用第一性原理 (FP) 计算和机器学习 (ML) 方法研究了元素。我们的 FP 计算表明 Hf、Ta 和 Ti 掺杂在热力学上是最稳定的。基于FP计算数据,开发了具有三种化学成分(CC)和中心环境(CE)特征的ML模型来预测Co 3的形成能和晶格常数(Al, X) (X = 3d、4d 和 5d TM 元素)。结果表明,这些 ML 模型都具有良好的预测精度,平均绝对误差 (<MAE > ) 为形成能~0.02 eV/原子,晶格常数为~0.01 Å。然后,讨论了重要特征对能量和几何特性的影响。为了研究合金化对 Co 3 (Al, W) 析出相稳定性的影响,发现Co 3 (Al, X)的 ML 模型同样准确地预测 Co 3 (Al, WX 3 )的未训练结构其中 X = 3d、4d 和 5d TM 元素,不包括 Co 和 W。我们发现 Co 3 (Al, WX 3) 结构在 X 是 IVB 和 VB 族 TM 元素时变得最稳定。这项工作表明,机器学习方法可以有效地扩展多组分合金设计中结构稳定性的第一性原理预测能力。








































 京公网安备 11010802027423号
京公网安备 11010802027423号