Kinetics and Catalysis ( IF 1.3 ) Pub Date : 2021-08-19 , DOI: 10.1134/s0023158421040108 S. V. Puchkov 1 , Yu. V. Nepomnyashchikh 1
|
Abstract
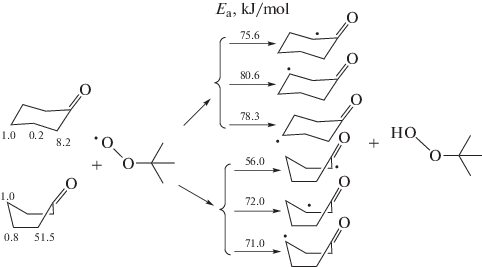
The effective charges on hydrogen and carbon atoms and the Fukui functions for the electrophilic and radical attacks for the hydrogen atoms of the chair and boat conformations of cyclohexanone were calculated using the density functional theory (DFT) with the hybrid functionals: B3LYP in the 6-311g++(d,p) basis set; X3LYP in the 6-311g++(d,p) and D95++(d,p) basis sets; and pbe1pbe in the D95++(d,p) basis set, and by the MP2 method in the 6-311g++(d,p) basis set. For these conformations, the transition states were localized; the length of the broken CH bond in the transition state was determined; and the activation energies, activation entropies, and enthalpies of the elementary reactions of the tert-butylperoxyl ((СН3)3СOO•) radical with all types of CH bonds of cyclohexanone were calculated. The relative reactivity of the CH bonds of cyclohexanone in reactions with ((СН3)3СOO•); the local electrophilicity indices of the carbon-centered radicals formed from cyclohexanone after elimination of the hydrogen atom; and the energy difference (ΔЕSOMO) between the single-occupied molecular orbitals of these carbon-centered radicals and the (СН3)3СOO• radical were calculated. The CH bond dissociation energies of cyclohexanone were calculated by the DFT B3LYP 6-311g++(d,p), CBS-QB3, and G3MP2B3 methods using the isodesmic reactions technique. The factors governing the CH bond reactivity of cyclohexanone in reactions with the (СН3)3СOO• radical were considered. The increased reactivity of CH bonds of cyclohexanone in the 2, 6 (α) positions was shown to be due to the low bond dissociation energy, high electron-donor capacity of hydrogen atoms, smaller length of the broken CH bond in the transition state, and small ΔЕSOMO value. The lower reactivity of CH bonds at positions 3, 5 (β), and 4 (γ) is determined by the higher bond dissociation energies, decreased electron density due to the inductive effect of the carbonyl group, nucleophilicity of the carbon-centered radicals, greater length of the broken CH bonds in the transition state, and larger ΔЕSOMO value. The difference in the reactivity of CH bonds at positions 3, 5 (β), and 4 (γ) is determined by these factors, but not by the difference in the dissociation energies of these bonds. The reactivity of CH bonds in the boat conformation is higher than that in the chair conformation in the reactions with the (СН3)3СOO• radical.
中文翻译:
用量子化学方法评价环己酮СН键在与叔丁基过氧自由基反应中的反应性
摘要
使用密度泛函理论 (DFT) 和混合泛函:6-中的 B3LYP 计算了氢和碳原子上的有效电荷以及对环己酮的椅子和船构象的氢原子进行亲电和自由基攻击的 Fukui 函数。 311g++( d , p )基组;X3LYP 在 6-311g++( d , p ) 和 D95++( d , p ) 基组中;和 pbe1pbe 在 D95++( d , p ) 基组中,通过 MP2 方法在 6-311g++( d , p) 基组。对于这些构象,过渡态是局部的;确定过渡态中断裂的 CH 键的长度;并计算了叔丁基过氧化氢((СН 3 ) 3 СOO • )自由基与环己酮的所有类型的CH键的基元反应的活化能、活化熵和焓。环己酮的 CH 键与 ((СН 3 ) 3 СOO • )反应的相对反应性;环己酮消除氢原子后形成的以碳为中心的自由基的局域亲电性指数;和能量差 (Δ Е SOMO) 在这些以碳为中心的自由基的单占据分子轨道和 (СН 3 ) 3 СOO •自由基之间进行了计算。环己酮的 CH 键解离能通过 DFT B3LYP 6-311g++( d , p )、CBS-QB3 和 G3MP2B3 方法使用等线反应技术计算。管理环己酮的CH键的反应性的反应中与(СН的因素3)3 СOO •激进被考虑。环己酮在 2, 6 (α) 位置的 CH 键的反应性增加被证明是由于键解离能低、氢原子的电子供体容量高、过渡态中断裂的 CH 键长度更短,和小ΔЕ SOMO值。CH 键在 3、5 (β) 和 4 (γ) 位置的较低反应性取决于较高的键解离能、由于羰基的诱导作用导致的电子密度降低、以碳为中心的自由基的亲核性,过渡态中断裂的 CH 键的长度越大,Δ Е SOMO越大价值。CH 键在 3、5 (β) 和 4 (γ) 位置的反应性差异由这些因素决定,而不是由这些键的解离能差异决定。在与 (СН 3 ) 3 СOO •自由基的反应中,船构象中的 CH 键的反应性高于椅子构象中的反应性。










































 京公网安备 11010802027423号
京公网安备 11010802027423号