当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Machine Learning Directed Optimization of Classical Molecular Modeling Force Fields
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2021-08-17 , DOI: 10.1021/acs.jcim.1c00448 Bridgette J Befort 1 , Ryan S DeFever 1 , Garrett M Tow 1 , Alexander W Dowling 1 , Edward J Maginn 1
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2021-08-17 , DOI: 10.1021/acs.jcim.1c00448 Bridgette J Befort 1 , Ryan S DeFever 1 , Garrett M Tow 1 , Alexander W Dowling 1 , Edward J Maginn 1
Affiliation
|
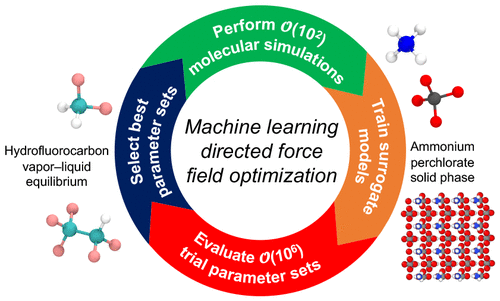
Accurate force fields are necessary for predictive molecular simulations. However, developing force fields that accurately reproduce experimental properties is challenging. Here, we present a machine learning directed, multiobjective optimization workflow for force field parametrization that evaluates millions of prospective force field parameter sets while requiring only a small fraction of them to be tested with molecular simulations. We demonstrate the generality of the approach and identify multiple low-error parameter sets for two distinct test cases: simulations of hydrofluorocarbon (HFC) vapor–liquid equilibrium (VLE) and an ammonium perchlorate (AP) crystal phase. We discuss the challenges and implications of our force field optimization workflow.
中文翻译:

经典分子建模力场的机器学习定向优化
预测性分子模拟需要精确的力场。然而,开发能够准确再现实验特性的力场具有挑战性。在这里,我们提出了一个用于力场参数化的机器学习导向的多目标优化工作流程,该工作流程评估数百万个预期的力场参数集,同时只需要使用分子模拟测试其中的一小部分。我们展示了该方法的通用性,并为两个不同的测试案例确定了多个低误差参数集:氢氟烃 (HFC) 气液平衡 (VLE) 和高氯酸铵 (AP) 晶相的模拟。我们讨论了力场优化工作流程的挑战和影响。
更新日期:2021-09-27
中文翻译:
经典分子建模力场的机器学习定向优化
预测性分子模拟需要精确的力场。然而,开发能够准确再现实验特性的力场具有挑战性。在这里,我们提出了一个用于力场参数化的机器学习导向的多目标优化工作流程,该工作流程评估数百万个预期的力场参数集,同时只需要使用分子模拟测试其中的一小部分。我们展示了该方法的通用性,并为两个不同的测试案例确定了多个低误差参数集:氢氟烃 (HFC) 气液平衡 (VLE) 和高氯酸铵 (AP) 晶相的模拟。我们讨论了力场优化工作流程的挑战和影响。










































 京公网安备 11010802027423号
京公网安备 11010802027423号