Journal of Physics and Chemistry of Solids ( IF 4 ) Pub Date : 2021-08-17 , DOI: 10.1016/j.jpcs.2021.110337 Francisco Colmenero 1 , Bruno Lunelli 2
|
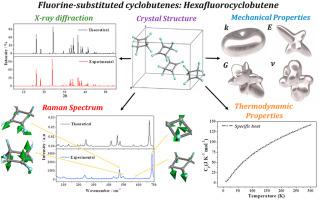
The crystal structures, infrared and Raman spectra, and mechanical and thermodynamic properties of four important fluorine-substituted cyclobutene derivatives in the solid state are investigated using first-principles solid-state methods based on density functional theory. These compounds are hexafluorocyclobutene (HFCB, ), 1,3,3,4,4-pentafluoro-2-methoxycyclobut-1-ene (PFMCB, ), 3,3,4,4-tetrafluoro-1,2-dimethoxycyclobut-1-ene [TFDMCB, and 1,2-dichloro-3,3,4,4-tetrafluorocyclobut-1-ene (DCTFCB, ). Although some of the properties of the corresponding molecules in the gas phase have been studied, and the structures of the corresponding molecular crystals have been determined by refinement from X-ray diffraction data, their vibrational spectra and properties have not yet been reported. The computed crystal structures and X-ray diffraction patterns are in excellent agreement with their experimental counterparts. The infrared and Raman spectra are calculated from the computed crystal structures using density functional perturbation theory. The results are highly consistent with the corresponding spectra measured experimentally in the gas or liquid phases and, therefore, appropriate normal coordinate analyses of the theoretical results are employed to rigorously assign all the vibrational bands. The elasticity matrices of these materials are computed using the finite deformation technique and a complete set of relevant mechanical properties is determined. Their equations of state are also obtained. These compounds are shown to be weak, highly anisotropic materials displaying the negative Poisson's ratio phenomenon. The TFDMCB also exhibits the negative linear compressibility (NLC) phenomenon for external isotropic pressures in the range of 0.64–1.76 GPa. The computed minimum compressibility, found at P = 0.73 GPa, is substantial (192.9 ). The NLC effect in TFDMCB is due to a collective rotation of the molecules within the crystal under increasing pressure. Finally, the thermodynamic properties of these materials are determined as a function of temperature using phonon calculations. The computed specific heats of HFCB, PFMCB, TFDMCB and DCMCB at T = 250 K are 127.5, 150.4, 169.9 and 134.8 , respectively, and corresponding entropies are 152.8, 173.3, 195.4 and 186.6 .
中文翻译:
固态氟取代环丁烯:晶体结构、振动光谱以及机械和热力学特性
使用基于密度泛函理论的第一性原理固态方法研究了固态中四种重要的氟取代环丁烯衍生物的晶体结构、红外和拉曼光谱以及机械和热力学性质。这些化合物是六氟环丁烯 (HFCB,), 1,3,3,4,4-五氟-2-甲氧基环丁-1-烯 (PFMCB, ), 3,3,4,4-四氟-1,2-二甲氧基环丁-1-烯 [TFDMCB, 和 1,2-二氯-3,3,4,4-四氟环丁-1-烯 (DCTFCB, )。尽管已经研究了气相中相应分子的一些性质,并且已经通过对 X 射线衍射数据的细化确定了相应分子晶体的结构,但它们的振动光谱和性质尚未得到报道。计算出的晶体结构和 X 射线衍射图与其实验对应物非常一致。红外和拉曼光谱是使用密度泛函微扰理论从计算出的晶体结构中计算出来的。结果与在气相或液相中实验测量的相应光谱高度一致,因此,采用理论结果的适当法向坐标分析来严格分配所有振动带。使用有限变形技术计算这些材料的弹性矩阵,并确定一整套相关的机械性能。也得到了它们的状态方程。这些化合物被证明是弱的、高度各向异性的材料,显示出负泊松比现象。对于 0.64-1.76 GPa 范围内的外部各向同性压力,TFDMCB 还表现出负线性压缩率 (NLC) 现象。在 P = 0.73 GPa 处发现的计算出的最小压缩率是相当大的(对于 0.64-1.76 GPa 范围内的外部各向同性压力,TFDMCB 还表现出负线性压缩率 (NLC) 现象。在 P = 0.73 GPa 处发现的计算出的最小压缩率是相当大的(对于 0.64-1.76 GPa 范围内的外部各向同性压力,TFDMCB 还表现出负线性压缩率 (NLC) 现象。在 P = 0.73 GPa 处发现的计算出的最小压缩率是相当大的(192.9 )。TFDMCB 中的 NLC 效应是由于在增加的压力下晶体内分子的集体旋转。最后,使用声子计算将这些材料的热力学特性确定为温度的函数。HFCB、PFMCB、TFDMCB 和 DCMCB 在 T = 250 K 时的计算比热分别为 127.5、150.4、169.9 和 134.8,分别对应的熵分别为 152.8、173.3、195.4 和 186.6 .


























 京公网安备 11010802027423号
京公网安备 11010802027423号