当前位置:
X-MOL 学术
›
Biochemistry
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Drugging the Undruggable: How Isoquinolines and PKA Initiated the Era of Designed Protein Kinase Inhibitor Therapeutics
Biochemistry ( IF 2.9 ) Pub Date : 2021-08-09 , DOI: 10.1021/acs.biochem.1c00359 Robin Lorenz 1 , Jian Wu 2 , Friedrich W Herberg 1 , Susan S Taylor 2, 3 , Richard A Engh 4
Biochemistry ( IF 2.9 ) Pub Date : 2021-08-09 , DOI: 10.1021/acs.biochem.1c00359 Robin Lorenz 1 , Jian Wu 2 , Friedrich W Herberg 1 , Susan S Taylor 2, 3 , Richard A Engh 4
Affiliation
|
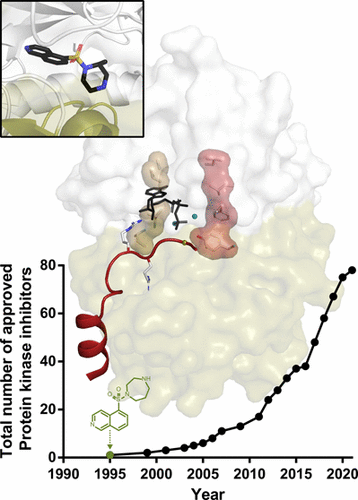
In 1984, Japanese researchers led by the biochemist Hiroyoshi Hidaka described the first synthetic protein kinase inhibitors based on an isoquinoline sulfonamide structure (Hidaka et al. Biochemistry, 1984 Oct 9; 23(21): 5036–41. doi: 10.1021/bi00316a032). These led to the first protein kinase inhibitor approved for medical use (fasudil), an inhibitor of the AGC subfamily Rho kinase. With potencies strong enough to compete against endogenous ATP, the isoquinoline compounds established the druggability of the ATP binding site. Crystal structures of their protein kinase complexes, including with cAMP-dependent protein kinase (PKA), showed interactions that, on the one hand, could mimic ATP but, on the other hand, could be optimized for high potency binding, kinase selectivity, and diversification away from adenosine. They also showed the flexibility of the glycine-rich loop, and PKA became a major prototype for crystallographic and nuclear magnetic resonance (NMR) studies of protein kinase mechanism and dynamic activity control. Since fasudil, more than 70 kinase inhibitors have been approved for clinical use, involving efforts that progressively have introduced new paradigms of data-driven drug discovery. Publicly available data alone comprise over 5000 protein kinase crystal structures and hundreds of thousands of binding data. Now, new methods, including artificial intelligence techniques and expansion of protein kinase targeting approaches, together with the expiration of patent protection for optimized inhibitor scaffolds, promise even greater advances in drug discovery. Looking back to the time of the first isoquinoline hinge binders brings the current state-of-the-art into stark contrast. Appropriately for this Perspective article, many of the milestone papers during this time were published in Biochemistry (now ACS Biochemistry).
中文翻译:

药物治疗:异喹啉和 PKA 如何开启设计蛋白激酶抑制剂治疗的时代
1984 年,由生物化学家 Hiroyoshi Hidaka 领导的日本研究人员描述了第一个基于异喹啉磺酰胺结构的合成蛋白激酶抑制剂(Hidaka 等,Biochemistry,1984 年 10 月 9 日;23(21):5036–41。doi:10.1021/bi032316a0a . 这些导致了第一个批准用于医疗用途的蛋白激酶抑制剂 (fasudil),一种 AGC 亚家族 Rho 激酶的抑制剂。由于具有足以与内源性 ATP 竞争的效力,异喹啉化合物确立了 ATP 结合位点的成药性。它们的蛋白激酶复合物的晶体结构,包括 cAMP 依赖性蛋白激酶 (PKA),显示出相互作用,一方面可以模拟 ATP,另一方面可以优化高效结合、激酶选择性和远离腺苷的多样化。他们还展示了富含甘氨酸的环的灵活性,PKA 成为蛋白质激酶机制和动态活性控制的晶体学和核磁共振 (NMR) 研究的主要原型。自法舒地尔以来,已有超过 70 种激酶抑制剂被批准用于临床,这些努力逐渐引入了数据驱动的药物发现新范式。仅公开可用的数据就包含超过 5000 个蛋白激酶晶体结构和数十万个结合数据。现在,包括人工智能技术和蛋白激酶靶向方法扩展在内的新方法,以及优化抑制剂支架专利保护的到期,有望在药物发现方面取得更大进展。回顾第一个异喹啉铰链粘合剂的时代,与当前最先进的技术形成鲜明对比。与这篇 Perspective 文章相吻合的是,这段时间的许多里程碑式论文都发表在 Biochemistry(现在的 ACS Biochemistry)上。
更新日期:2021-08-09
中文翻译:
药物治疗:异喹啉和 PKA 如何开启设计蛋白激酶抑制剂治疗的时代
1984 年,由生物化学家 Hiroyoshi Hidaka 领导的日本研究人员描述了第一个基于异喹啉磺酰胺结构的合成蛋白激酶抑制剂(Hidaka 等,Biochemistry,1984 年 10 月 9 日;23(21):5036–41。doi:10.1021/bi032316a0a . 这些导致了第一个批准用于医疗用途的蛋白激酶抑制剂 (fasudil),一种 AGC 亚家族 Rho 激酶的抑制剂。由于具有足以与内源性 ATP 竞争的效力,异喹啉化合物确立了 ATP 结合位点的成药性。它们的蛋白激酶复合物的晶体结构,包括 cAMP 依赖性蛋白激酶 (PKA),显示出相互作用,一方面可以模拟 ATP,另一方面可以优化高效结合、激酶选择性和远离腺苷的多样化。他们还展示了富含甘氨酸的环的灵活性,PKA 成为蛋白质激酶机制和动态活性控制的晶体学和核磁共振 (NMR) 研究的主要原型。自法舒地尔以来,已有超过 70 种激酶抑制剂被批准用于临床,这些努力逐渐引入了数据驱动的药物发现新范式。仅公开可用的数据就包含超过 5000 个蛋白激酶晶体结构和数十万个结合数据。现在,包括人工智能技术和蛋白激酶靶向方法扩展在内的新方法,以及优化抑制剂支架专利保护的到期,有望在药物发现方面取得更大进展。回顾第一个异喹啉铰链粘合剂的时代,与当前最先进的技术形成鲜明对比。与这篇 Perspective 文章相吻合的是,这段时间的许多里程碑式论文都发表在 Biochemistry(现在的 ACS Biochemistry)上。










































 京公网安备 11010802027423号
京公网安备 11010802027423号