当前位置:
X-MOL 学术
›
ACS Chem. Biol.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
A Comparison of Two Stability Proteomics Methods for Drug Target Identification in OnePot 2D Format
ACS Chemical Biology ( IF 3.5 ) Pub Date : 2021-08-10 , DOI: 10.1021/acschembio.1c00317 Yingrong Xu 1 , Graham M West 1 , Mario Abdelmessih 1 , Matthew D Troutman 1 , Robert A Everley 1
ACS Chemical Biology ( IF 3.5 ) Pub Date : 2021-08-10 , DOI: 10.1021/acschembio.1c00317 Yingrong Xu 1 , Graham M West 1 , Mario Abdelmessih 1 , Matthew D Troutman 1 , Robert A Everley 1
Affiliation
|
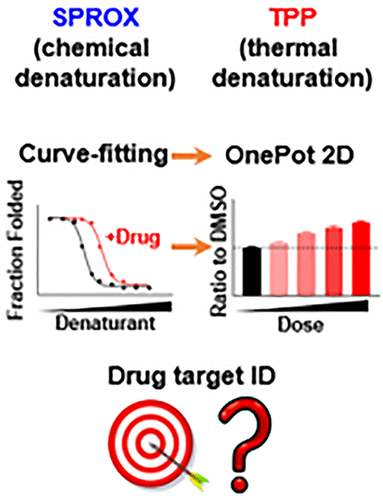
Stability proteomics techniques that do not require drug modifications have emerged as an attractive alternative to affinity purification methods in drug target engagement studies. Two representative techniques include the chemical-denaturation-based SPROX (Stability of Proteins from Rates of Oxidation), which utilizes peptide-level quantification and thermal-denaturation-based TPP (Thermal Proteome Profiling), which utilizes protein-level quantification. Recently, the “OnePot” strategy was adapted for both SPROX and TPP to increase the throughput. When combined with the 2D setup which measures both the denaturation and the drug dose dimensions, the OnePot 2D format offers improved analysis specificity with higher resource efficiency. However, a systematic evaluation of the OnePot 2D format and a comparison between SPROX and TPP are still lacking. Here, we performed SPROX and TPP to identify protein targets of a well-studied pan-kinase inhibitor staurosporine with K562 lysate, in curve-fitting and OnePot 2D formats. We found that the OnePot 2D format provided ∼10× throughput, achieved ∼1.6× protein coverage and involves more straightforward data analysis. We also compared SPROX with the current “gold-standard” stability proteomics technique TPP in the OnePot 2D format. The protein coverage of TPP is ∼1.5 fold of SPROX; however, SPROX offers protein domain-level information, identifies comparable numbers of kinase hits, has higher signal (R value), and requires ∼3× less MS time. Unique SPROX hits encompass higher-molecular-weight proteins, compared to the unique TPP hits, and include atypical kinases. We also discuss hit stratification and prioritization strategies to promote the efficiency of hit followup.
中文翻译:

OnePot 2D 格式药物靶标鉴定的两种稳定性蛋白质组学方法的比较
不需要药物修饰的稳定性蛋白质组学技术已成为药物靶标研究中亲和纯化方法的一种有吸引力的替代方法。两种代表性技术包括基于化学变性的 SPROX(来自氧化速率的蛋白质稳定性),它利用肽级量化和基于热变性的 TPP(热蛋白质组分析),它利用蛋白质级量化。最近,SPROX 和 TPP 都采用了“OnePot”策略以提高吞吐量。当与测量变性和药物剂量维度的 2D 设置相结合时,OnePot 2D 格式提供了改进的分析特异性和更高的资源效率。然而,仍然缺乏对 OnePot 2D 格式的系统评估以及 SPROX 和 TPP 之间的比较。在这里,我们执行了 SPROX 和 TPP,以曲线拟合和 OnePot 2D 格式识别经过充分研究的泛激酶抑制剂星形孢菌素与 K562 裂解物的蛋白质靶标。我们发现 OnePot 2D 格式提供了约 10 倍的吞吐量,实现了约 1.6 倍的蛋白质覆盖率,并且涉及更直接的数据分析。我们还将 SPROX 与 OnePot 2D 格式的当前“黄金标准”稳定性蛋白质组学技术 TPP 进行了比较。TPP 的蛋白质覆盖率是 SPROX 的 1.5 倍;然而,SPROX 提供蛋白质域级别的信息,识别可比数量的激酶命中,具有更高的信号(R 值),并且需要的 MS 时间减少约 3 倍。与独特的 TPP 命中相比,独特的 SPROX 命中包含更高分子量的蛋白质,并包括非典型激酶。
更新日期:2021-08-20
中文翻译:
OnePot 2D 格式药物靶标鉴定的两种稳定性蛋白质组学方法的比较
不需要药物修饰的稳定性蛋白质组学技术已成为药物靶标研究中亲和纯化方法的一种有吸引力的替代方法。两种代表性技术包括基于化学变性的 SPROX(来自氧化速率的蛋白质稳定性),它利用肽级量化和基于热变性的 TPP(热蛋白质组分析),它利用蛋白质级量化。最近,SPROX 和 TPP 都采用了“OnePot”策略以提高吞吐量。当与测量变性和药物剂量维度的 2D 设置相结合时,OnePot 2D 格式提供了改进的分析特异性和更高的资源效率。然而,仍然缺乏对 OnePot 2D 格式的系统评估以及 SPROX 和 TPP 之间的比较。在这里,我们执行了 SPROX 和 TPP,以曲线拟合和 OnePot 2D 格式识别经过充分研究的泛激酶抑制剂星形孢菌素与 K562 裂解物的蛋白质靶标。我们发现 OnePot 2D 格式提供了约 10 倍的吞吐量,实现了约 1.6 倍的蛋白质覆盖率,并且涉及更直接的数据分析。我们还将 SPROX 与 OnePot 2D 格式的当前“黄金标准”稳定性蛋白质组学技术 TPP 进行了比较。TPP 的蛋白质覆盖率是 SPROX 的 1.5 倍;然而,SPROX 提供蛋白质域级别的信息,识别可比数量的激酶命中,具有更高的信号(R 值),并且需要的 MS 时间减少约 3 倍。与独特的 TPP 命中相比,独特的 SPROX 命中包含更高分子量的蛋白质,并包括非典型激酶。








































 京公网安备 11010802027423号
京公网安备 11010802027423号