当前位置:
X-MOL 学术
›
Phys. Status Solidi. Rapid Res. Lett.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
High-Throughput Electronic Structures and Ferroelectric Interfaces of HfO2 by GGA+U(d,p) Calculations
Physica Status Solidi-Rapid Research Letters ( IF 2.5 ) Pub Date : 2021-07-27 , DOI: 10.1002/pssr.202100295 Yuzheng Guo 1 , Zhaofu Zhang 2 , John Robertson 2
Physica Status Solidi-Rapid Research Letters ( IF 2.5 ) Pub Date : 2021-07-27 , DOI: 10.1002/pssr.202100295 Yuzheng Guo 1 , Zhaofu Zhang 2 , John Robertson 2
Affiliation
|
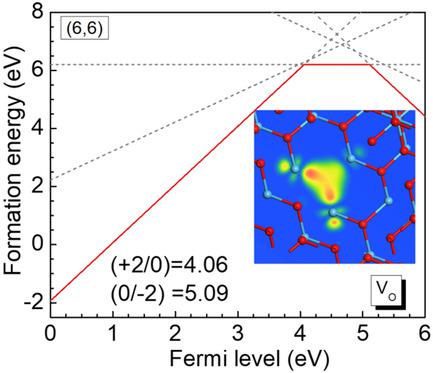
The electronic structure, vacancy symmetry, defect levels, ferroelectric phases, and interface properties of HfO2 are studied using a GGA + U(d,p) approach, a simplified version of the ACBN0 method. Introducing an on-site Coulomb interaction to both Hf 5d orbitals and O 2p orbitals reproduces the experimental bandgap, gives band energies similar to those of hybrid functionals, gives the correct symmetry for the oxygen vacancy, and describes the Schottky barriers at the metallic contacts like TiN correctly. The energetics of phase energies and strain arising from different ferroelectric–electrode interfaces are tested. The GGA + U(d,p) approach is a useful tool to study various HfO2 configurations by rapid ab initio molecular dynamics calculations.
中文翻译:

通过 GGA+U(d,p) 计算得到 HfO2 的高通量电子结构和铁电界面
使用 GGA + U ( d, p ) 方法(ACBN0 方法的简化版本)研究 HfO 2的电子结构、空位对称性、缺陷能级、铁电相和界面特性。将现场库仑相互作用引入 Hf 5 d轨道和 O 2 p轨道,重现了实验带隙,提供了类似于杂化泛函的能带能量,给出了氧空位的正确对称性,并描述了金属上的肖特基势垒正确接触像 TiN。测试了不同铁电-电极界面产生的相能能量和应变。GGA + U ( d,p) 方法是通过快速从头算分子动力学计算研究各种 HfO 2构型的有用工具。
更新日期:2021-07-27
中文翻译:
通过 GGA+U(d,p) 计算得到 HfO2 的高通量电子结构和铁电界面
使用 GGA + U ( d, p ) 方法(ACBN0 方法的简化版本)研究 HfO 2的电子结构、空位对称性、缺陷能级、铁电相和界面特性。将现场库仑相互作用引入 Hf 5 d轨道和 O 2 p轨道,重现了实验带隙,提供了类似于杂化泛函的能带能量,给出了氧空位的正确对称性,并描述了金属上的肖特基势垒正确接触像 TiN。测试了不同铁电-电极界面产生的相能能量和应变。GGA + U ( d,p) 方法是通过快速从头算分子动力学计算研究各种 HfO 2构型的有用工具。










































 京公网安备 11010802027423号
京公网安备 11010802027423号