当前位置:
X-MOL 学术
›
Phys. Status Solidi. Rapid Res. Lett.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Effects of Bromine Substitution and Vacancy Defects on the Structural and Electronic Properties of Black Orthorhombic CsPbI3 Perovskite
Physica Status Solidi-Rapid Research Letters ( IF 2.5 ) Pub Date : 2021-07-27 , DOI: 10.1002/pssr.202100277 Zhonghai Lin 1 , Jiayi Lei 1 , Pingjian Wang 1 , Ling Xu 1 , Xiaoxiao Zhang 1 , Yunxin Kang 1 , Mingyu Chen 1 , Guangfen Wei 1
Physica Status Solidi-Rapid Research Letters ( IF 2.5 ) Pub Date : 2021-07-27 , DOI: 10.1002/pssr.202100277 Zhonghai Lin 1 , Jiayi Lei 1 , Pingjian Wang 1 , Ling Xu 1 , Xiaoxiao Zhang 1 , Yunxin Kang 1 , Mingyu Chen 1 , Guangfen Wei 1
Affiliation
|
The structural and electronic properties of both Br substitution and vacancy structures of black orthorhombic CsPbI3 (γ-CsPbI3) perovskite are investigated by carrying out first-principle calculations in density functional theory (DFT). For mixed perovskites CsPb(I1−x
Br
x
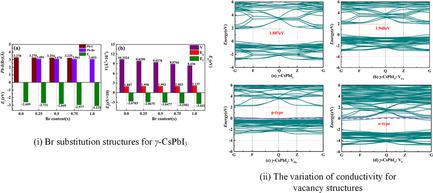
)3, x is 0.0, 0.25, 0.5, 0.75, and 1.0, respectively. The studies suggest that the III site should be preferentially substituted by Br atom rather than the II site and the structure becomes more stable with increasing Br content. These compounds are direct-bandgap semiconductors in the range of 1.887−2.137 eV. Moreover, the vacancy changes the electroconductibility of γ-CsPbI3. γ-CsPbI3:VPb and γ-CsPbI3:VI exhibit p-type and n-type conductivity, respectively. γ-CsPbI3:VCs still is a semiconductor with a direct bandgap, which presents a slight decrease in electroconductibility due to the increase in bandgap. The calculated structural parameters show that both substitution and vacancy can induce structural distortion. Partial density of states (PDOS) suggests that the top of the valence band arises from hybridization of Pb s- and halogen p-orbitals, whereas the bottom of the conduction band has predominantly Pb p-orbitals for two kinds of crystal structures. These results provide strong support for developing high-performance perovskite photovoltaic materials in optoelectronic devices.
中文翻译:

溴取代和空位缺陷对黑色正交晶系 CsPbI3 钙钛矿结构和电子特性的影响
通过在密度泛函理论 (DFT) 中进行第一性原理计算,研究了黑色斜方晶 CsPbI 3 (γ - CsPbI 3 ) 钙钛矿的Br 取代和空位结构的结构和电子性质。对于混合钙钛矿 CsPb(I 1− x Br x ) 3,x 分别为0.0、0.25、0.5、0.75和 1.0。研究表明,III位点应该优先被 Br 原子取代,而不是 I I 位点,结构随着 Br 含量的增加而变得更加稳定。这些化合物是 1.887-2.137 eV 范围内的直接带隙半导体。此外,空位改变了γ-CsPbI 3的导电性。γ-CsPbI 3 :V Pb和γ-CsPbI 3 :V I 分别表现出p型和n型导电性。γ-CsPbI 3 :V Cs仍然是具有直接带隙的半导体,由于带隙的增加,其导电性略有下降。计算的结构参数表明取代和空位都可以引起结构变形。部分态密度 (PDOS) 表明价带顶部来自 Pb s - 和卤素p -轨道的杂化,而导带底部主要具有两种晶体结构的Pb p -轨道。这些结果为在光电器件中开发高性能钙钛矿光伏材料提供了强有力的支持。
更新日期:2021-07-27
中文翻译:
溴取代和空位缺陷对黑色正交晶系 CsPbI3 钙钛矿结构和电子特性的影响
通过在密度泛函理论 (DFT) 中进行第一性原理计算,研究了黑色斜方晶 CsPbI 3 (γ - CsPbI 3 ) 钙钛矿的Br 取代和空位结构的结构和电子性质。对于混合钙钛矿 CsPb(I 1− x Br x ) 3,x 分别为0.0、0.25、0.5、0.75和 1.0。研究表明,III位点应该优先被 Br 原子取代,而不是 I I 位点,结构随着 Br 含量的增加而变得更加稳定。这些化合物是 1.887-2.137 eV 范围内的直接带隙半导体。此外,空位改变了γ-CsPbI 3的导电性。γ-CsPbI 3 :V Pb和γ-CsPbI 3 :V I 分别表现出p型和n型导电性。γ-CsPbI 3 :V Cs仍然是具有直接带隙的半导体,由于带隙的增加,其导电性略有下降。计算的结构参数表明取代和空位都可以引起结构变形。部分态密度 (PDOS) 表明价带顶部来自 Pb s - 和卤素p -轨道的杂化,而导带底部主要具有两种晶体结构的Pb p -轨道。这些结果为在光电器件中开发高性能钙钛矿光伏材料提供了强有力的支持。










































 京公网安备 11010802027423号
京公网安备 11010802027423号