当前位置:
X-MOL 学术
›
Clin. Transl. Immunol.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Dilemmas in the diagnosis and pathogenesis of atypical late-onset familial haemophagocytic lymphohistiocytosis
Clinical & Translational Immunology ( IF 4.6 ) Pub Date : 2021-07-26 , DOI: 10.1002/cti2.1320 Adrian Minson 1, 2 , Ilia Voskoboinik 2 , Andrew Grigg 1
Clinical & Translational Immunology ( IF 4.6 ) Pub Date : 2021-07-26 , DOI: 10.1002/cti2.1320 Adrian Minson 1, 2 , Ilia Voskoboinik 2 , Andrew Grigg 1
Affiliation
|
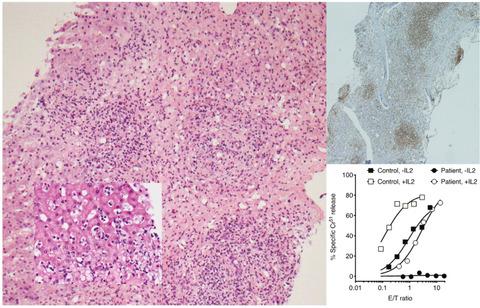
A congenital loss of cytotoxic lymphocyte activity leads to a potentially fatal immune dysregulation, familial haemophagocytic lymphohistiocytosis. Until recently, this disease was uniformly associated with infants or very young children, but it appears now that the onset may be delayed for decades. As a result, some adults are being mis- or under-diagnosed because of their ‘atypical’ symptoms that are not recognised as immunodeficiency. The clinical picture and histopathology can overlap with those of haematologic malignancy, further complicating the diagnostic thought process. The spectrum of atypical symptoms is poorly defined, and therefore, it is important to describe these cases and the attendant immunological and cellular changes associated with familial haemophagocytic lymphohistiocytosis, in order to improve diagnosis and prevent unintended consequences of symptomatic therapies.
中文翻译:

非典型迟发性家族性噬血细胞性淋巴组织细胞增多症诊断及发病机制的困境
先天性细胞毒性淋巴细胞活性的丧失导致潜在的致命免疫失调,即家族性噬血细胞性淋巴组织细胞增多症。直到最近,这种疾病一直与婴儿或非常年幼的儿童有关,但现在看来,发病可能会延迟数十年。结果,一些成年人因为不被认为是免疫缺陷的“非典型”症状而被误诊或诊断不足。临床表现和组织病理学可能与血液系统恶性肿瘤重叠,使诊断思维过程更加复杂。非典型症状的范围定义不明确,因此,重要的是描述这些病例以及与家族性噬血细胞性淋巴组织细胞增多症相关的免疫学和细胞变化,
更新日期:2021-07-26
中文翻译:
非典型迟发性家族性噬血细胞性淋巴组织细胞增多症诊断及发病机制的困境
先天性细胞毒性淋巴细胞活性的丧失导致潜在的致命免疫失调,即家族性噬血细胞性淋巴组织细胞增多症。直到最近,这种疾病一直与婴儿或非常年幼的儿童有关,但现在看来,发病可能会延迟数十年。结果,一些成年人因为不被认为是免疫缺陷的“非典型”症状而被误诊或诊断不足。临床表现和组织病理学可能与血液系统恶性肿瘤重叠,使诊断思维过程更加复杂。非典型症状的范围定义不明确,因此,重要的是描述这些病例以及与家族性噬血细胞性淋巴组织细胞增多症相关的免疫学和细胞变化,










































 京公网安备 11010802027423号
京公网安备 11010802027423号