Surface Science ( IF 2.1 ) Pub Date : 2021-07-23 , DOI: 10.1016/j.susc.2021.121904 Kun'ichi Miyazawa 1 , Yumi Tanaka 1
|
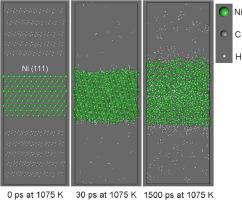
The activation energy of methane decomposition by nickel was calculated using models of nickel thin films, a large-scale atomic/molecular massively parallel simulator (LAMMPS) and a ReaxFF potential. The rate constants of the first-order chemical reaction of methane decomposition were obtained by analyzing the number of undecomposed methane molecules in individual movie frames. The obtained activation energies of methane decomposition on the Ni (111) surfaces showed values close to those reported in the literature. The structural change of a Ni thin film with (111) surfaces was simulated to investigate how carbon and hydrogen atoms are deposited on the Ni thin film upon the decomposition of methane molecules at a high temperature. The catalytic activity of the (111) Ni thin film was found to decrease from the time point of critical supersaturation of carbon in the Ni thin film.
中文翻译:
高温下镍薄膜上甲烷分解的 LAMMPS 分子动力学模拟
使用镍薄膜模型、大规模原子/分子大规模并行模拟器 (LAMMPS) 和 ReaxFF 电位计算镍分解甲烷的活化能。甲烷分解一级化学反应的速率常数是通过分析单个电影帧中未分解甲烷分子的数量获得的。在 Ni(111)表面上获得的甲烷分解活化能显示出接近文献报道的值。模拟具有(111)面的Ni薄膜的结构变化,以研究在高温下甲烷分子分解时碳和氢原子如何沉积在Ni薄膜上。










































 京公网安备 11010802027423号
京公网安备 11010802027423号