当前位置:
X-MOL 学术
›
Phys. Rev. Materials
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Enhanced interlayer interactions in Ni-dopedMoS2, and structural and electronic signatures of doping site
Physical Review Materials ( IF 3.1 ) Pub Date : 2021-07-23 , DOI: 10.1103/physrevmaterials.5.074006 Rijan Karkee , Enrique Guerrero , David A. Strubbe
Physical Review Materials ( IF 3.1 ) Pub Date : 2021-07-23 , DOI: 10.1103/physrevmaterials.5.074006 Rijan Karkee , Enrique Guerrero , David A. Strubbe
|
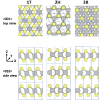
The crystal structure of with strong covalent bonds in plane and weak van der Waals interactions out of plane gives rise to interesting properties for applications such as solid lubrication, optoelectronics, and catalysis, which can be enhanced by transition-metal doping. However, the mechanisms for improvement and even the structure of the doped material can be unclear, which we address with theoretical calculations. Building on our previous work on Ni doping of the bulk 2H phase, now we compare to polytypes (1H monolayer and 3R bulk), to determine favorable sites for Ni and the doping effect on structure, electronic properties, and the layer dissociation energy. The most favorable intercalation or adatom sites are tetrahedral intercalation for 3R (like 2H) and Mo atop for 1H. The relative energies indicate a possibility of phase change from 2H to 3R with substitution of Mo or S. We find structural and electronic properties that can be used to identify the doping sites, including metallic behavior in Mo-substituted 3R and 2H, and in-gap states for Mo- and S-substituted 1H, which could have interesting optoelectronic applications. We observe a large enhancement in the interlayer interactions of Ni-doped , opposite to the effect of other transition metals. For lubrication applications, this increased layer dissociation energy could be the mechanism of low wear. Our systematic study shows the effect of doping concentration and we extrapolate to the low-doping limit. This work gives insight into the previously unclear structure of Ni-doped and how it can be detected experimentally, the relation of energy and structures of doped monolayers and bulk systems, the electronic properties under doping, and the effect of doping on interlayer interactions.
中文翻译:

Ni掺杂MoS2中增强的层间相互作用,以及掺杂位点的结构和电子特征
的晶体结构 平面内强共价键和平面外弱范德华相互作用产生了有趣的特性,可用于固体润滑、光电子和催化等应用,这些特性可以通过过渡金属掺杂得到增强。然而,掺杂材料的改进机制甚至结构都不清楚,我们通过理论计算来解决。基于我们之前对本体 2H 相的 Ni 掺杂所做的工作,现在我们与多型体(1H 单层和 3R 本体)进行比较,以确定 Ni 的有利位点以及掺杂对结构、电子特性和层离解能的影响。最有利的嵌入或吸附原子位点是 3R(如 2H)的四面体嵌入和 1H 的 Mo 顶部。相对能量表明了从 2H 到 3R 的相变的可能性,其中取代了 Mo 或 S。我们发现了可用于识别掺杂位点的结构和电子特性,包括 Mo 取代的 3R 和 2H 中的金属行为,以及Mo 和 S 取代的 1H 的间隙态,这可能具有有趣的光电应用。我们观察到 Ni 掺杂的层间相互作用有很大的增强,与其他过渡金属的作用相反。对于润滑应用,这种增加的层离解能可能是低磨损的机制。我们的系统研究显示了掺杂浓度的影响,我们推断出低掺杂极限。这项工作使人们深入了解了以前不清楚的 Ni 掺杂结构 以及如何通过实验检测它,掺杂单层和体系统的能量和结构的关系,掺杂下的电子特性,以及掺杂对层间相互作用的影响。
更新日期:2021-07-23
中文翻译:
Ni掺杂MoS2中增强的层间相互作用,以及掺杂位点的结构和电子特征
的晶体结构 平面内强共价键和平面外弱范德华相互作用产生了有趣的特性,可用于固体润滑、光电子和催化等应用,这些特性可以通过过渡金属掺杂得到增强。然而,掺杂材料的改进机制甚至结构都不清楚,我们通过理论计算来解决。基于我们之前对本体 2H 相的 Ni 掺杂所做的工作,现在我们与多型体(1H 单层和 3R 本体)进行比较,以确定 Ni 的有利位点以及掺杂对结构、电子特性和层离解能的影响。最有利的嵌入或吸附原子位点是 3R(如 2H)的四面体嵌入和 1H 的 Mo 顶部。相对能量表明了从 2H 到 3R 的相变的可能性,其中取代了 Mo 或 S。我们发现了可用于识别掺杂位点的结构和电子特性,包括 Mo 取代的 3R 和 2H 中的金属行为,以及Mo 和 S 取代的 1H 的间隙态,这可能具有有趣的光电应用。我们观察到 Ni 掺杂的层间相互作用有很大的增强,与其他过渡金属的作用相反。对于润滑应用,这种增加的层离解能可能是低磨损的机制。我们的系统研究显示了掺杂浓度的影响,我们推断出低掺杂极限。这项工作使人们深入了解了以前不清楚的 Ni 掺杂结构 以及如何通过实验检测它,掺杂单层和体系统的能量和结构的关系,掺杂下的电子特性,以及掺杂对层间相互作用的影响。










































 京公网安备 11010802027423号
京公网安备 11010802027423号