当前位置:
X-MOL 学术
›
Acta Cryst. B
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Developing orbital-free quantum crystallography: the local potentials and associated partial charge densities
Acta Crystallographica Section B ( IF 2.684 ) Pub Date : 2021-07-19 , DOI: 10.1107/s2052520621005540 Vladimir Tsirelson , Adam Stash
Acta Crystallographica Section B ( IF 2.684 ) Pub Date : 2021-07-19 , DOI: 10.1107/s2052520621005540 Vladimir Tsirelson , Adam Stash
|
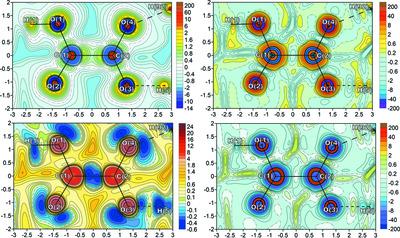
This work extends the orbital-free density functional theory to the field of quantum crystallography. The total electronic energy is decomposed into electrostatic, exchange, Weizsacker and Pauli components on the basis of physically grounded arguments. Then, the one-electron Euler equation is re-written through corresponding potentials, which have clear physical and chemical meaning. Partial electron densities related with these potentials by the Poisson equation are also defined. All these functions were analyzed from viewpoint of their physical content and limits of applicability. Then, they were expressed in terms of experimental electron density and its derivatives using the orbital-free density functional theory approximations, and applied to the study of chemical bonding in a heteromolecular crystal of ammonium hydrooxalate oxalic acid dihydrate. It is demonstrated that this approach allows the electron density to be decomposed into physically meaningful components associated with electrostatics, exchange, and spin-independent wave properties of electrons or with their combinations in a crystal. Therefore, the bonding information about a crystal that was previously unavailable for X-ray diffraction analysis can be now obtained.
中文翻译:

开发无轨道量子晶体学:局部电位和相关的部分电荷密度
这项工作将无轨道密度泛函理论扩展到量子晶体学领域。总电子能量在物理接地论证的基础上分解为静电、交换、魏茨萨克和泡利分量。然后,通过相应的势将一电子欧拉方程改写,具有明确的物理和化学意义。还通过泊松方程定义了与这些电位相关的部分电子密度。所有这些功能都从它们的物理内容和适用范围的角度进行了分析。然后,它们使用无轨道密度泛函理论近似以实验电子密度及其导数表示,并应用于草酸二水合草酸铵异分子晶体中化学键的研究。已经证明,这种方法允许将电子密度分解为与电子的静电、交换和与自旋无关的波特性或它们在晶体中的组合相关的物理上有意义的分量。因此,现在可以获得以前无法用于 X 射线衍射分析的晶体的键合信息。
更新日期:2021-08-05
中文翻译:
开发无轨道量子晶体学:局部电位和相关的部分电荷密度
这项工作将无轨道密度泛函理论扩展到量子晶体学领域。总电子能量在物理接地论证的基础上分解为静电、交换、魏茨萨克和泡利分量。然后,通过相应的势将一电子欧拉方程改写,具有明确的物理和化学意义。还通过泊松方程定义了与这些电位相关的部分电子密度。所有这些功能都从它们的物理内容和适用范围的角度进行了分析。然后,它们使用无轨道密度泛函理论近似以实验电子密度及其导数表示,并应用于草酸二水合草酸铵异分子晶体中化学键的研究。已经证明,这种方法允许将电子密度分解为与电子的静电、交换和与自旋无关的波特性或它们在晶体中的组合相关的物理上有意义的分量。因此,现在可以获得以前无法用于 X 射线衍射分析的晶体的键合信息。


























 京公网安备 11010802027423号
京公网安备 11010802027423号