当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Efficient workflow for the investigation of the catalytic cycle of water oxidation catalysts: Combining GFN-xTB and density functional theory
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2021-07-18 , DOI: 10.1002/jcc.26721 Jan Paul Menzel 1 , Martijn Kloppenburg 1 , Jelena Belić 2 , Huub J M de Groot 1 , Lucas Visscher 2 , Francesco Buda 1
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2021-07-18 , DOI: 10.1002/jcc.26721 Jan Paul Menzel 1 , Martijn Kloppenburg 1 , Jelena Belić 2 , Huub J M de Groot 1 , Lucas Visscher 2 , Francesco Buda 1
Affiliation
|
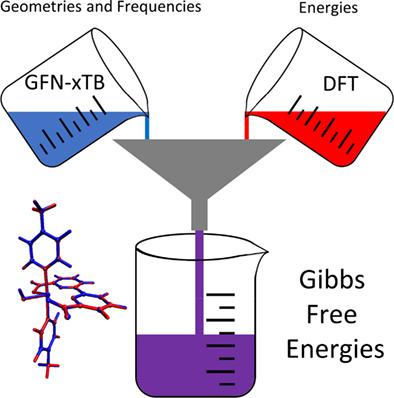
Photocatalytic water oxidation remains the bottleneck in many artificial photosynthesis devices. The efficiency of this challenging process is inherently linked to the thermodynamic and electronic properties of the chromophore and the water oxidation catalyst (WOC). Computational investigations can facilitate the search for favorable chromophore-catalyst combinations. However, this remains a demanding task due to the requirements on the computational method that should be able to correctly describe different spin and oxidation states of the transition metal, the influence of solvation and the different rates of the charge transfer and water oxidation processes. To determine a suitable method with favorable cost/accuracy ratios, the full catalytic cycle of a molecular ruthenium based WOC is investigated using different computational methods, including density functional theory (DFT) with different functionals (GGA, Hybrid, Double Hybrid) as well as the semi-empirical tight binding approach GFN-xTB. A workflow with low computational cost is proposed that combines GFN-xTB and DFT and provides reliable results. GFN-xTB geometries and frequencies combined with single-point DFT energies give free energy changes along the catalytic cycle that closely follow the full DFT results and show satisfactory agreement with experiment, while significantly decreasing the computational cost. This workflow allows for cost efficient determination of energetic, thermodynamic and dynamic properties of WOCs.
中文翻译:

研究水氧化催化剂催化循环的高效工作流程:结合 GFN-xTB 和密度泛函理论
光催化水氧化仍然是许多人工光合作用装置的瓶颈。这一具有挑战性的过程的效率与生色团和水氧化催化剂 (WOC) 的热力学和电子特性有着内在的联系。计算研究可以促进寻找有利的发色团催化剂组合。然而,由于对计算方法的要求应该能够正确描述过渡金属的不同自旋和氧化态、溶剂化的影响以及电荷转移和水氧化过程的不同速率,这仍然是一项艰巨的任务。为了确定具有有利成本/精度比的合适方法,使用不同的计算方法研究了基于分子钌的 WOC 的完整催化循环,包括具有不同泛函(GGA、Hybrid、Double Hybrid)的密度泛函理论 (DFT) 以及半经验紧束缚方法 GFN-xTB。提出了一种结合 GFN-xTB 和 DFT 并提供可靠结果的低计算成本的工作流程。GFN-xTB 几何形状和频率与单点 DFT 能量相结合,沿催化循环产生自由能变化,与完整的 DFT 结果密切相关,并与实验显示出令人满意的一致性,同时显着降低了计算成本。该工作流程允许经济高效地确定 WOC 的能量、热力学和动态特性。提出了一种结合 GFN-xTB 和 DFT 并提供可靠结果的低计算成本的工作流程。GFN-xTB 几何形状和频率与单点 DFT 能量相结合,沿催化循环产生自由能变化,与完整的 DFT 结果密切相关,并与实验显示出令人满意的一致性,同时显着降低了计算成本。该工作流程允许经济高效地确定 WOC 的能量、热力学和动态特性。提出了一种结合 GFN-xTB 和 DFT 并提供可靠结果的低计算成本的工作流程。GFN-xTB 几何形状和频率与单点 DFT 能量相结合,沿催化循环产生自由能变化,与完整的 DFT 结果密切相关,并与实验显示出令人满意的一致性,同时显着降低了计算成本。该工作流程允许经济高效地确定 WOC 的能量、热力学和动态特性。
更新日期:2021-08-15
中文翻译:
研究水氧化催化剂催化循环的高效工作流程:结合 GFN-xTB 和密度泛函理论
光催化水氧化仍然是许多人工光合作用装置的瓶颈。这一具有挑战性的过程的效率与生色团和水氧化催化剂 (WOC) 的热力学和电子特性有着内在的联系。计算研究可以促进寻找有利的发色团催化剂组合。然而,由于对计算方法的要求应该能够正确描述过渡金属的不同自旋和氧化态、溶剂化的影响以及电荷转移和水氧化过程的不同速率,这仍然是一项艰巨的任务。为了确定具有有利成本/精度比的合适方法,使用不同的计算方法研究了基于分子钌的 WOC 的完整催化循环,包括具有不同泛函(GGA、Hybrid、Double Hybrid)的密度泛函理论 (DFT) 以及半经验紧束缚方法 GFN-xTB。提出了一种结合 GFN-xTB 和 DFT 并提供可靠结果的低计算成本的工作流程。GFN-xTB 几何形状和频率与单点 DFT 能量相结合,沿催化循环产生自由能变化,与完整的 DFT 结果密切相关,并与实验显示出令人满意的一致性,同时显着降低了计算成本。该工作流程允许经济高效地确定 WOC 的能量、热力学和动态特性。提出了一种结合 GFN-xTB 和 DFT 并提供可靠结果的低计算成本的工作流程。GFN-xTB 几何形状和频率与单点 DFT 能量相结合,沿催化循环产生自由能变化,与完整的 DFT 结果密切相关,并与实验显示出令人满意的一致性,同时显着降低了计算成本。该工作流程允许经济高效地确定 WOC 的能量、热力学和动态特性。提出了一种结合 GFN-xTB 和 DFT 并提供可靠结果的低计算成本的工作流程。GFN-xTB 几何形状和频率与单点 DFT 能量相结合,沿催化循环产生自由能变化,与完整的 DFT 结果密切相关,并与实验显示出令人满意的一致性,同时显着降低了计算成本。该工作流程允许经济高效地确定 WOC 的能量、热力学和动态特性。


























 京公网安备 11010802027423号
京公网安备 11010802027423号